

Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori
- PMID: 22647547
- PMCID: PMC3429542
- DOI: 10.4161/auto.20159
Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori
Abstract
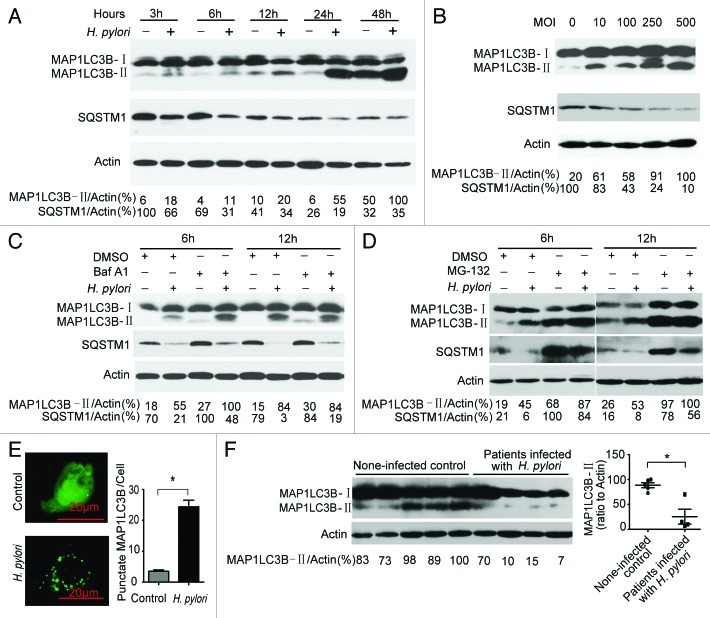
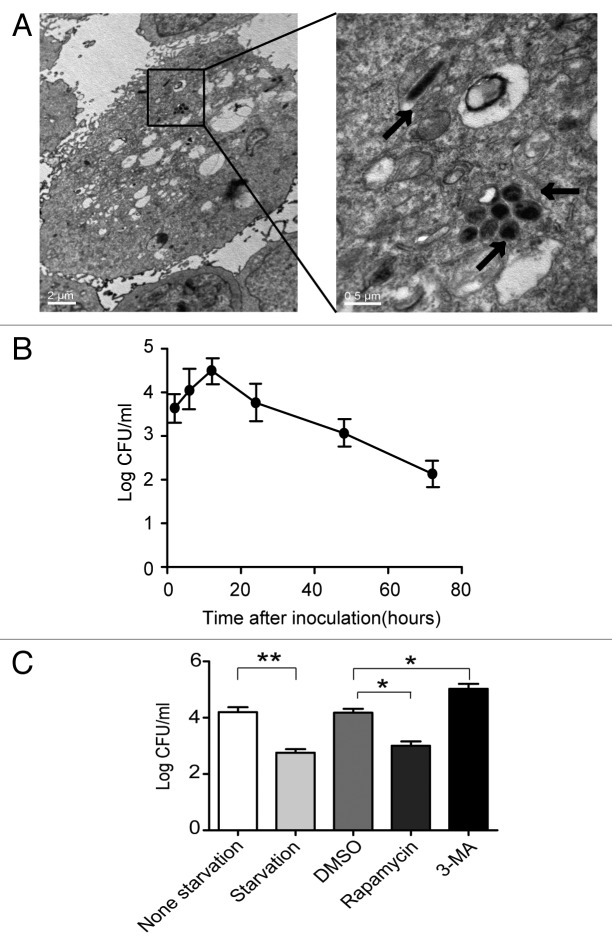
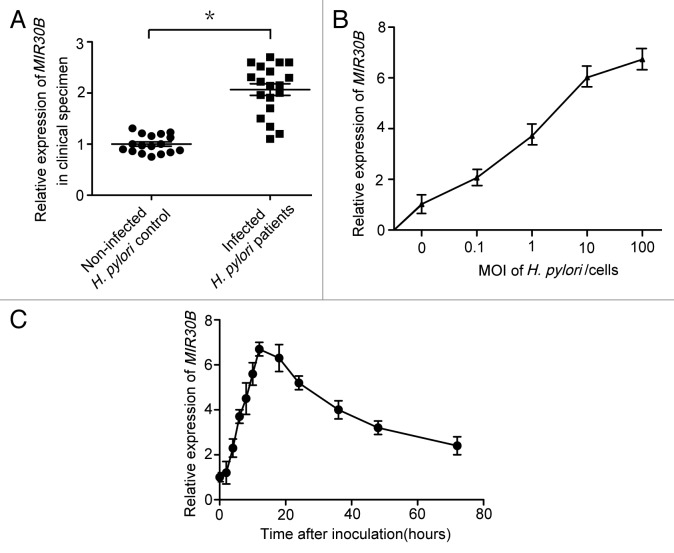
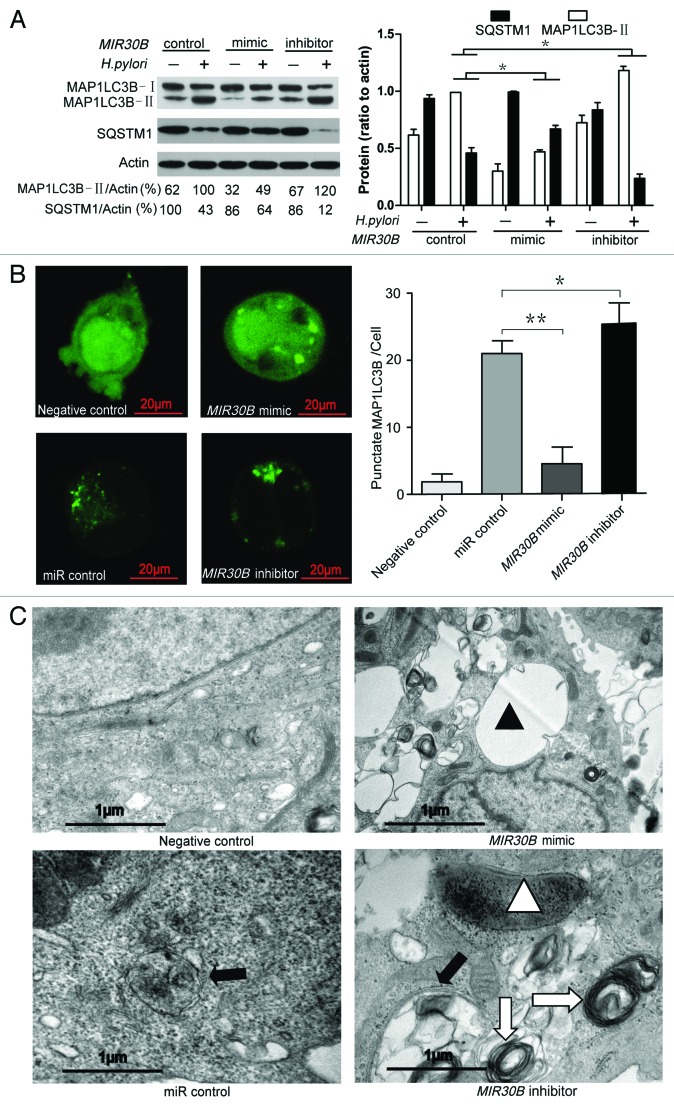
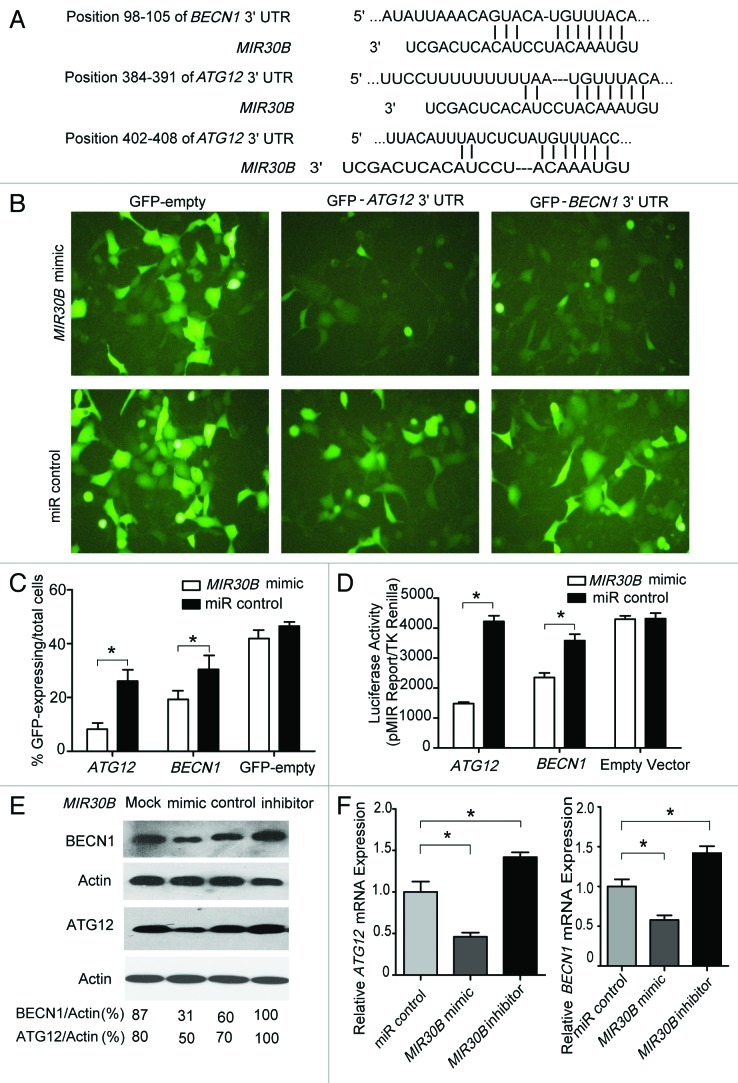
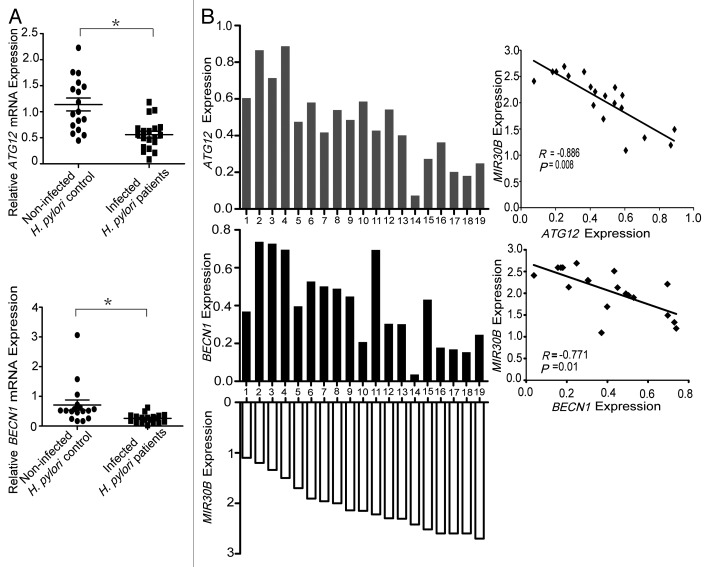
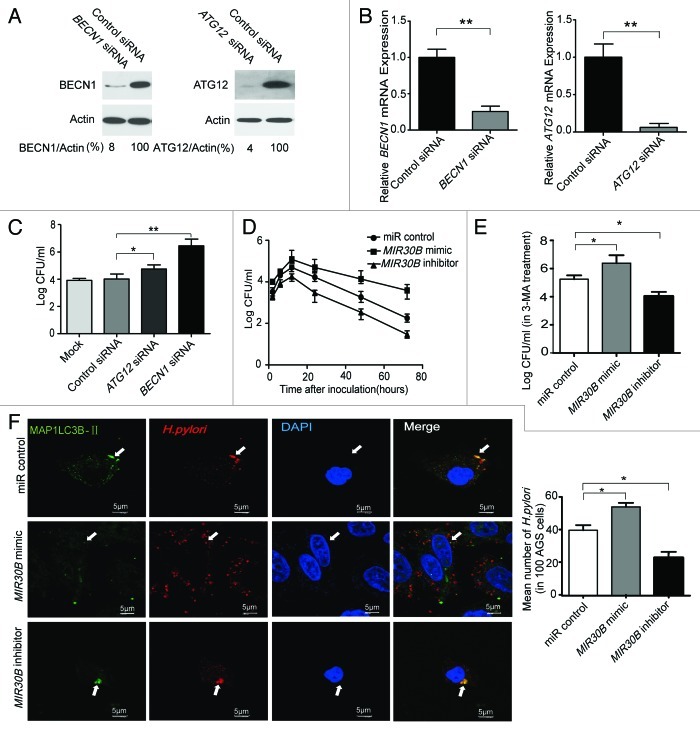
Helicobacter pylori evade immune responses and achieve persistent colonization in the stomach. However, the mechanism by which H. pylori infections persist is not clear. In this study, we showed that MIR30B is upregulated during H. pylori infection of an AGS cell line and human gastric tissues. Upregulation of MIR30B benefited bacterial replication by compromising the process of autophagy during the H. pylori infection. As a potential mechanistic explanation for this observation, we demonstrate that MIR30B directly targets ATG12 and BECN1, which are important proteins involved in autophagy. These results suggest that compromise of autophagy by MIR30B allows intracellular H. pylori to evade autophagic clearance, thereby contributing to the persistence of H. pylori infections.
Keywords: MIR30B; ATG12; BECN1; Helicobacter pylori; autophagy.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials