

Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia
- PMID: 22647877
- PMCID: PMC3393041
- DOI: 10.1161/CIRCRESAHA.112.270041
Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia
Abstract
Rationale: Ca binding to the troponin complex represents a major portion of cytosolic Ca buffering. Troponin mutations that increase myofilament Ca sensitivity are associated with familial hypertrophic cardiomyopathy and confer a high risk for sudden death. In mice, Ca sensitization causes ventricular arrhythmias, but the underlying mechanisms remain unclear.
Objective: To test the hypothesis that myofilament Ca sensitization increases cytosolic Ca buffering and to determine the resulting arrhythmogenic changes in Ca homeostasis in the intact mouse heart.
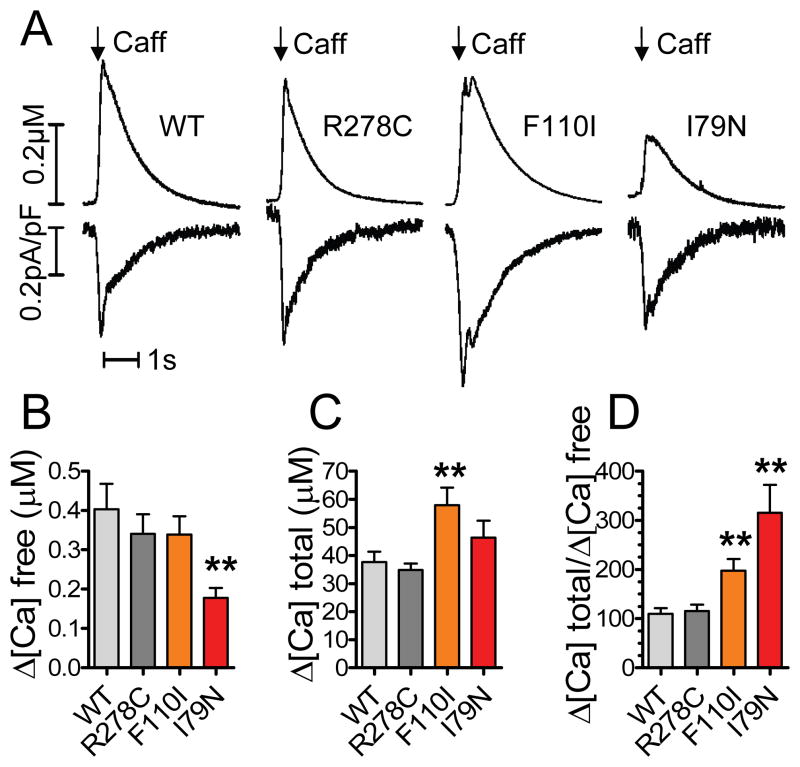
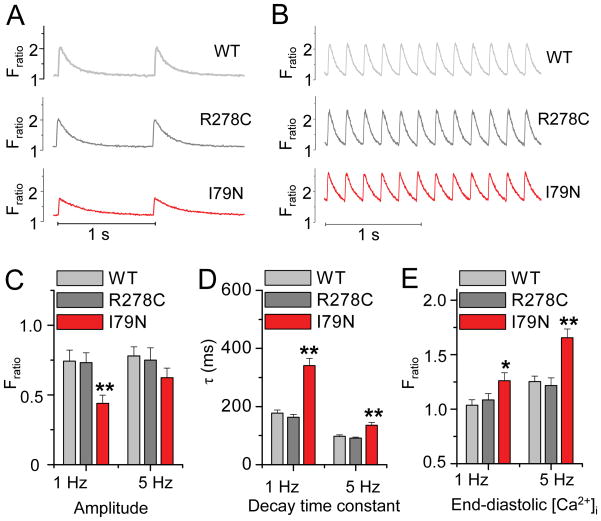
Methods and results: Using cardiomyocytes isolated from mice expressing troponin T (TnT) mutants (TnT-I79N, TnT-F110I, TnT-R278C), we found that increasing myofilament Ca sensitivity produced a proportional increase in cytosolic Ca binding. The underlying cause was an increase in the cytosolic Ca binding affinity, whereas maximal Ca binding capacity was unchanged. The effect was sufficiently large to alter Ca handling in intact mouse hearts at physiological heart rates, resulting in increased end-diastolic [Ca] at fast pacing rates, and enhanced sarcoplasmic reticulum Ca content and release after pauses. Accordingly, action potential (AP) regulation was altered, with postpause action potential prolongation, afterdepolarizations, and triggered activity. Acute Ca sensitization with EMD 57033 mimicked the effects of Ca-sensitizing TnT mutants and produced pause-dependent ventricular ectopy and sustained ventricular tachycardia after acute myocardial infarction.
Conclusions: Myofilament Ca sensitization increases cytosolic Ca binding affinity. A major proarrhythmic consequence is a pause-dependent potentiation of Ca release, action potential prolongation, and triggered activity. Increased cytosolic Ca binding represents a novel mechanism of pause-dependent arrhythmia that may be relevant for inherited and acquired cardiomyopathies.
Figures






References
-
- Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation. 2000;102(8):858–864. - PubMed
-
- Maron BJ, Shen WK, Link MS, Epstein AE, Almquist AK, Daubert JP, Bardy GH, Favale S, Rea RF, Boriani G, Estes NA, 3rd, Spirito P. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy [see comments] N Engl J Med. 2000;342(6):365–373. - PubMed
-
- Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342(24):1778–1785. - PubMed
-
- Varnava AM, Elliott PM, Baboonian C, Davison F, Davies MJ, McKenna WJ. Hypertrophic cardiomyopathy: histopathological features of sudden death in cardiac troponin T disease. Circulation. 2001;104(12):1380–1384. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
