

Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging
- PMID: 22649061
- PMCID: PMC3458524
- DOI: 10.1093/nar/gks454
Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging
Abstract
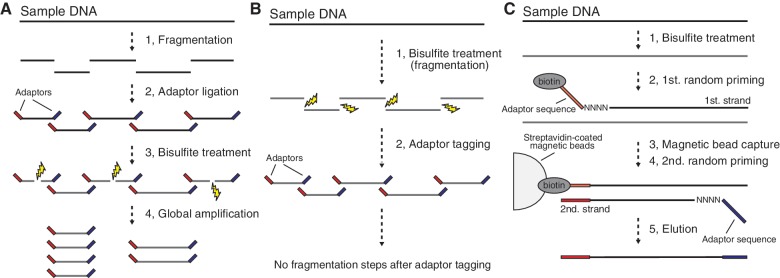
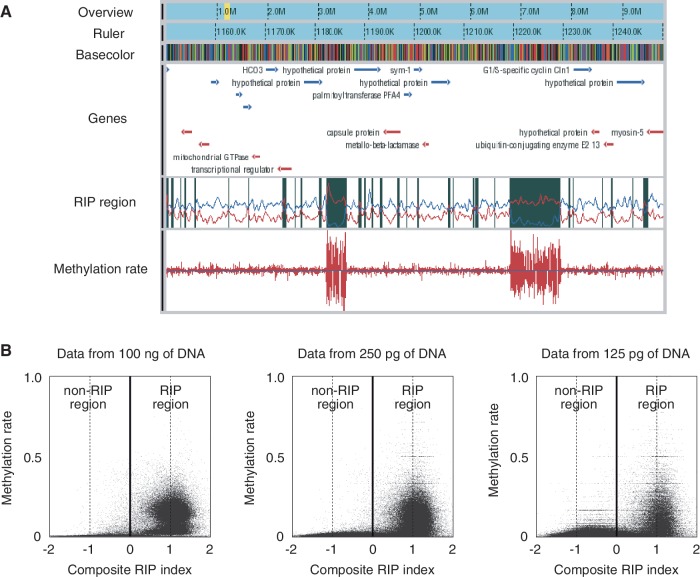
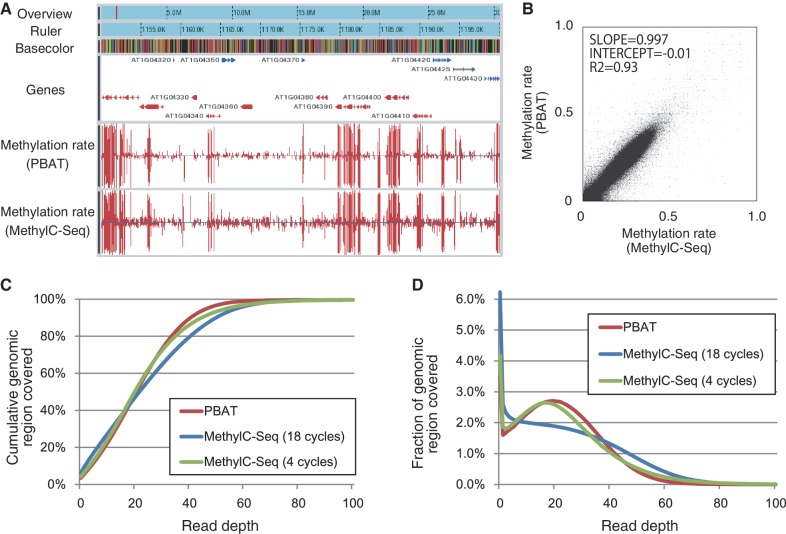
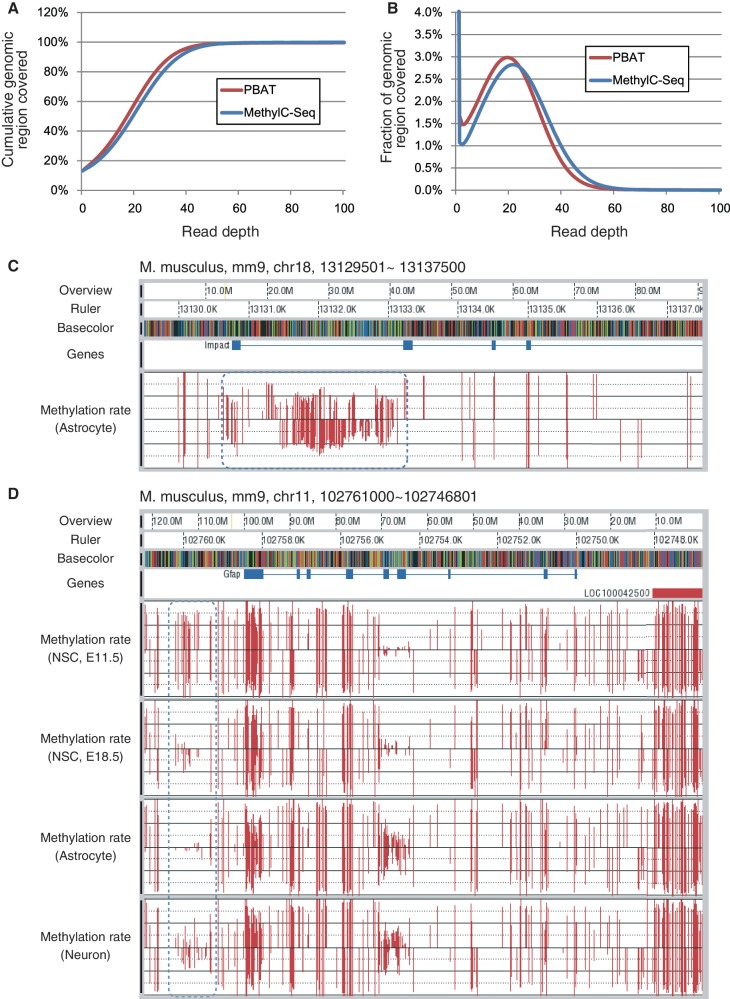
DNA methylation plays a key role in epigenetic regulation of eukaryotic genomes. Hence the genome-wide distribution of 5-methylcytosine, or the methylome, has been attracting intense attention. In recent years, whole-genome bisulfite sequencing (WGBS) has enabled methylome analysis at single-base resolution. However, WGBS typically requires microgram quantities of DNA as well as global PCR amplification, thereby precluding its application to samples of limited amounts. This is presumably because bisulfite treatment of adaptor-tagged templates, which is inherent to current WGBS methods, leads to substantial DNA fragmentation. To circumvent the bisulfite-induced loss of intact sequencing templates, we conceived an alternative method termed Post-Bisulfite Adaptor Tagging (PBAT) wherein bisulfite treatment precedes adaptor tagging by two rounds of random primer extension. The PBAT method can generate a substantial number of unamplified reads from as little as subnanogram quantities of DNA. It requires only 100 ng of DNA for amplification-free WGBS of mammalian genomes. Thus, the PBAT method will enable various novel applications that would not otherwise be possible, thereby contributing to the rapidly growing field of epigenomics.
Figures
References
-
- Xiang H, Zhu J, Chen Q, Dai F, Li X, Li M, Zhang H, Zhang G, Li D, Dong Y, et al. Single base-resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010;28:516–520. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
