

Extracellular domain N-glycosylation controls human thrombopoietin receptor cell surface levels
- PMID: 22649382
- PMCID: PMC3355985
- DOI: 10.3389/fendo.2011.00071
Extracellular domain N-glycosylation controls human thrombopoietin receptor cell surface levels
Abstract
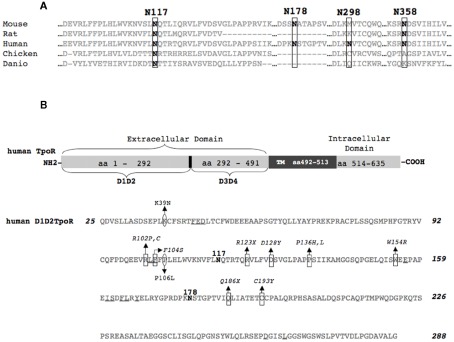
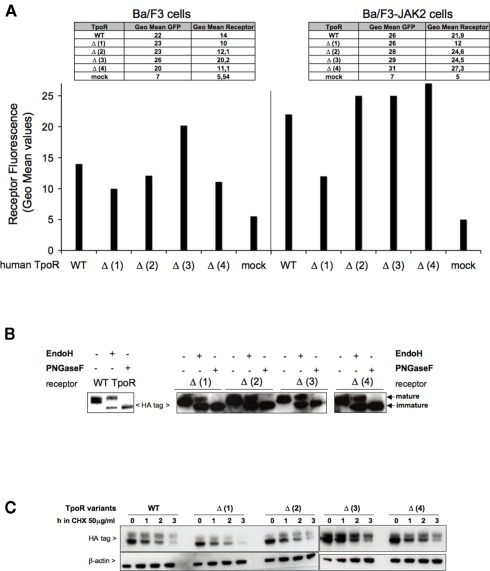
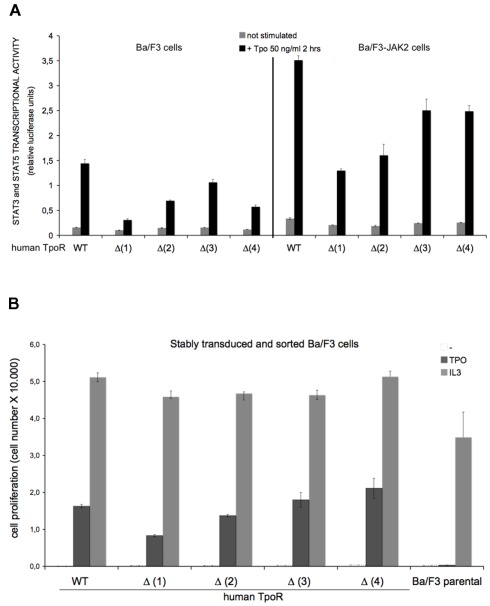
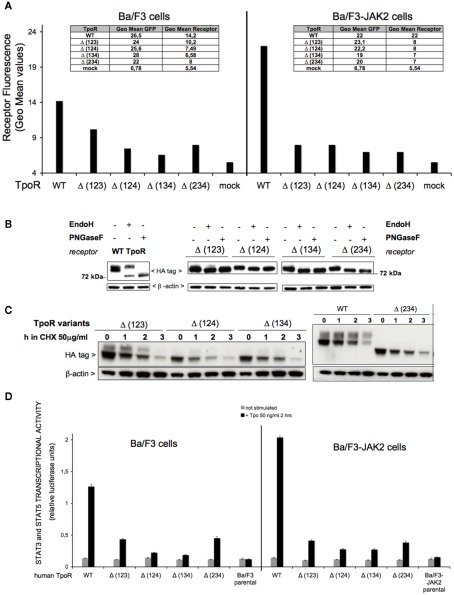
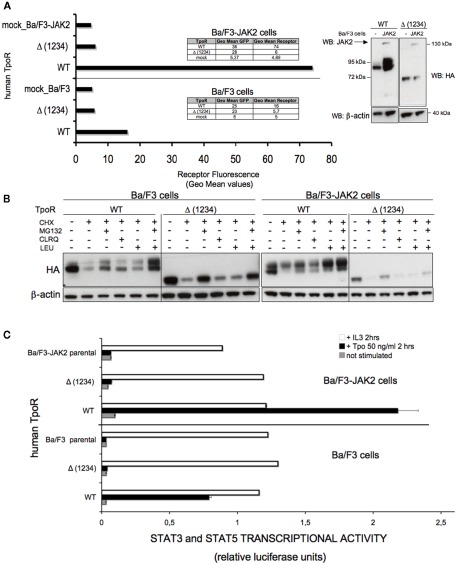
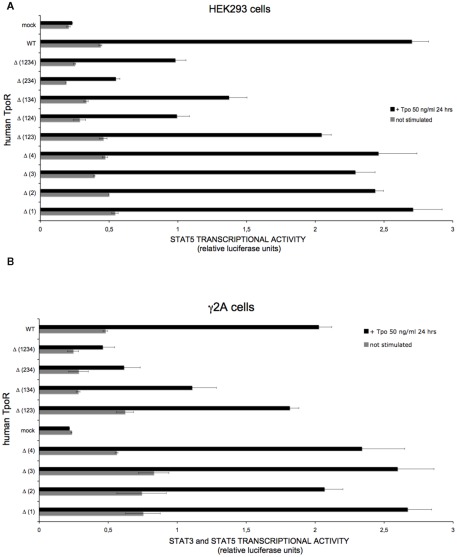
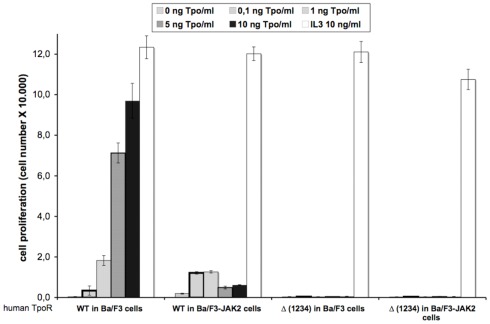
The thrombopoietin receptor (TpoR) is a type I transmembrane protein that mediates the signaling functions of thrombopoietin (Tpo) in regulating megakaryocyte differentiation, platelet formation, and hematopoietic stem cell renewal. We probed the role of each of the four extracellular domain putative N-glycosylation sites for cell surface localization and function of the receptor. Single N-glycosylation mutants at any of the four sites were able to acquire the mature N-glycosylated pattern, but exhibited a decreased Tpo-dependent JAK2-STAT response in stably transduced Ba/F3 or Ba/F3-JAK2 cell lines. The ability of JAK2 to promote cell surface localization and stability of TpoR required the first N-glycosylation site (Asn117). In contrast, the third N-glycosylation site (Asn298) decreased receptor maturation and stability. TpoR mutants lacking three N-glycosylation sites were defective in maturation, but N-glycosylation on the single remaining site could be detected by sensitivity to PNGaseF. The TpoR mutant defective in all four N-glycosylation sites was severely impaired in plasma membrane localization and was degraded by the proteasome. N-glycosylation receptor mutants are not misfolded as, once localized on the cell surface in overexpression conditions, they can bind and respond to Tpo. Our data indicate that extracellular domain N-glycosylation sites regulate in a combinatorial manner cell surface localization of TpoR. We discuss how mutations around TpoR N-glycosylation sites might contribute to inefficient receptor traffic and disease.
Keywords: ER maturation; JAK2; N-glycosylation; cell surface traffic; cytokine receptor; endoglycosidase H; signal transduction; thrombopoietin.
Figures
References
-
- Dalal I., Arpaia E., Dadi H., Kulkarni S., Squire J., Roifman C. M. (1998). Cloning and characterization of the human homolog of mouse Jak2. Blood 91, 844–851 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous