

Calcium signaling and neurodegeneration
- PMID: 22649630
- PMCID: PMC3347543
Calcium signaling and neurodegeneration
Abstract
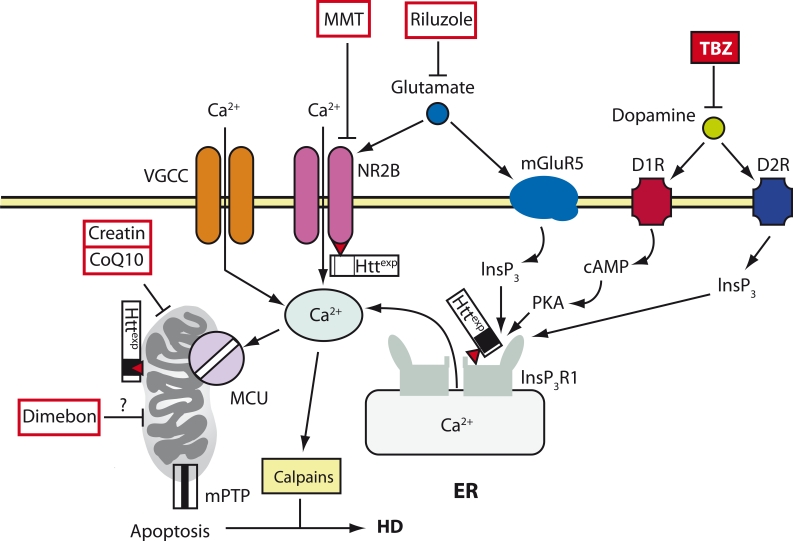
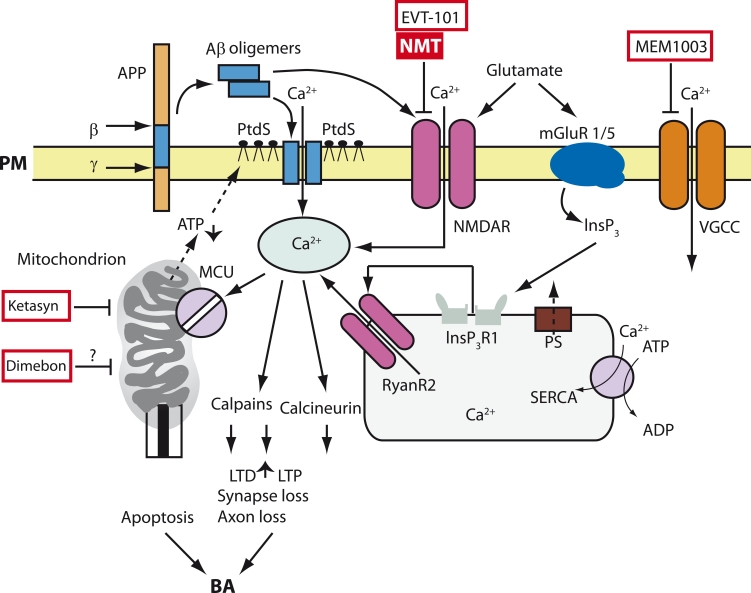
Neurodegenerative disorders, such as Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), and spinocerebellar ataxias (SCA) are very important both for fundamental science and for practical medicine. Despite extensive research into the causes of these diseases, clinical researchers have had very limited progress and, as of now, there is still no cure for any of these diseases. One of the main obstacles in the way of creating treatments for these disorders is the fact that their etiology and pathophysiology still remain unclear. This paper reviews results that support the so-called "calcium hypothesis of neurodegenerative diseases." The calcium hypothesis states that the atrophic and degenerative processes in the neurons of AD, PD, ALS, HD, and SCA patients are accompanied by alterations in calcium homeostasis. Moreover, the calcium hypothesis states that this deregulation of calcium signaling is one of the early-stage and key processes in the pathogenesis of these diseases. Based on the results we reviewed, we conclude that the calcium channels and other proteins involved in the neuronal calcium signaling system are potential drug targets for AD, PD, ALS, HD, and SCA therapy.
Keywords: Alzheimer’s disease; Huntington’s disease; Parkinson’s disease (PD); amyotrophic lateral sclerosis; calcium channels; calcium signaling; clinical trials; dimebon; imaging; memantine; mitochondria; riluzole; spinocerbellar ataxias; transgenic mice.
Figures
References
-
- Berridge M.J.. Neuronal Calcium Signaling. Neuron. 1998;21:13–26. - PubMed
-
- Toescu E.C, Verkhratsky A. . The Importance of Being Subtle: Small Changes in Calcium Homeostasis Control Cognitive Decline in Normal Aging. Aging Cell. 2007;6:267–273. - PubMed
-
- Foster T.C.. Calcium Homeostasis and Modulation of Synaptic Plasticity in the Aged Brain. Aging Cell. 2007;6:319–325. - PubMed
-
- Gusella J.F., MacDonald M.E.. Molecular Genetics: Unmasking Polyglutamine Triggers in Neurodegenerative Disease. Nat Rev Neurosci. 2000;1:109–115. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous