

Glycines: role in α-helical membrane protein structures and a potential indicator of native conformation
- PMID: 22650985
- PMCID: PMC3426646
- DOI: 10.1021/bi300090x
Glycines: role in α-helical membrane protein structures and a potential indicator of native conformation
Abstract
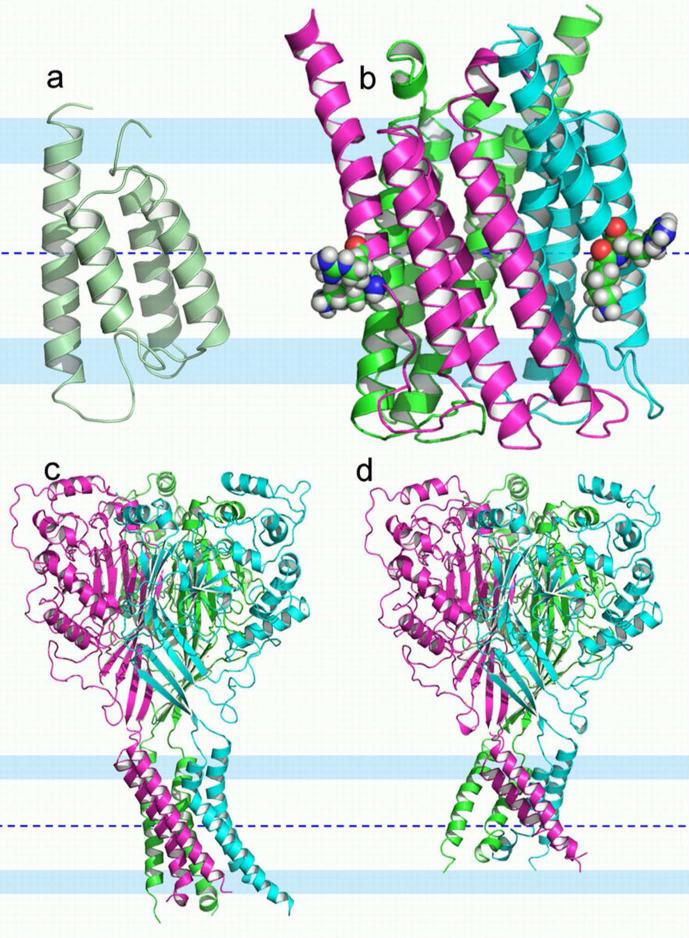
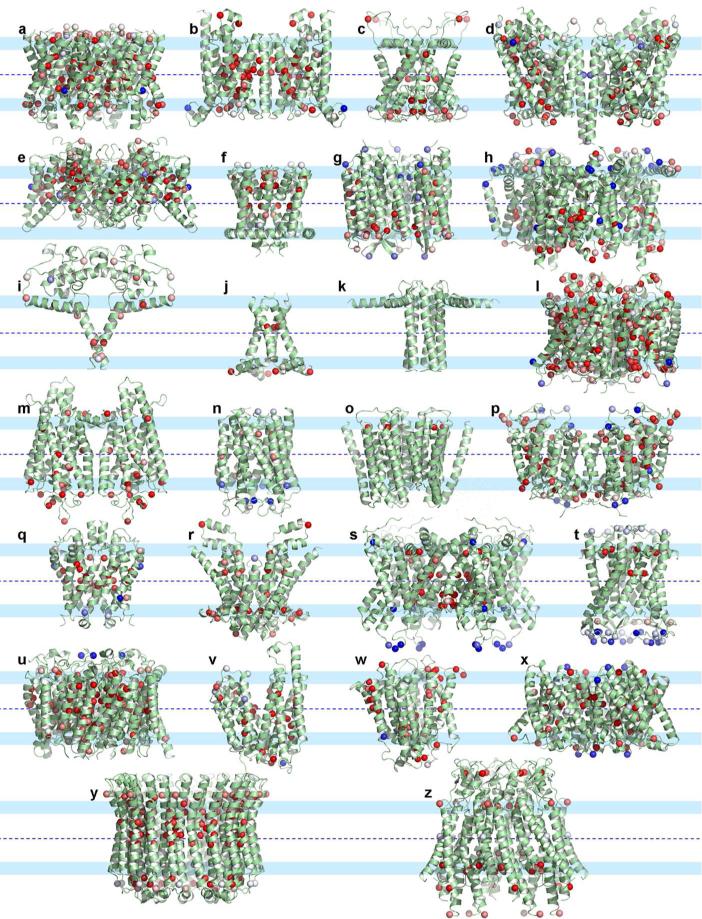
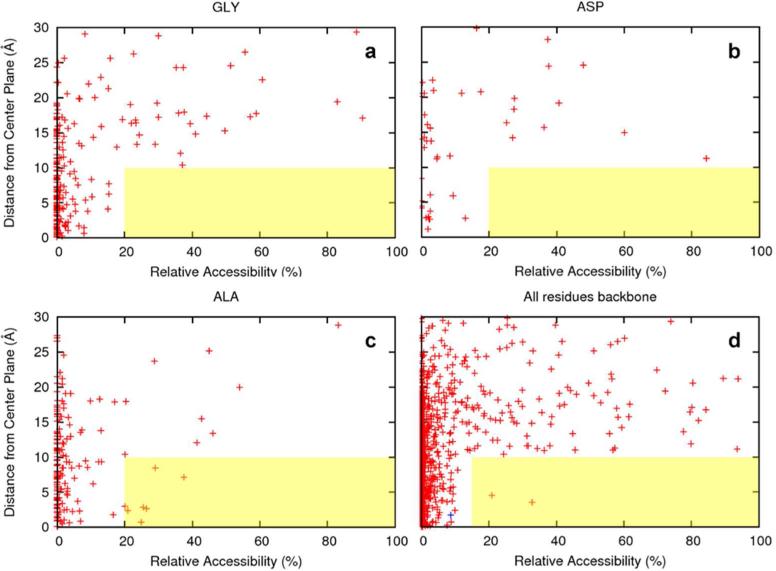
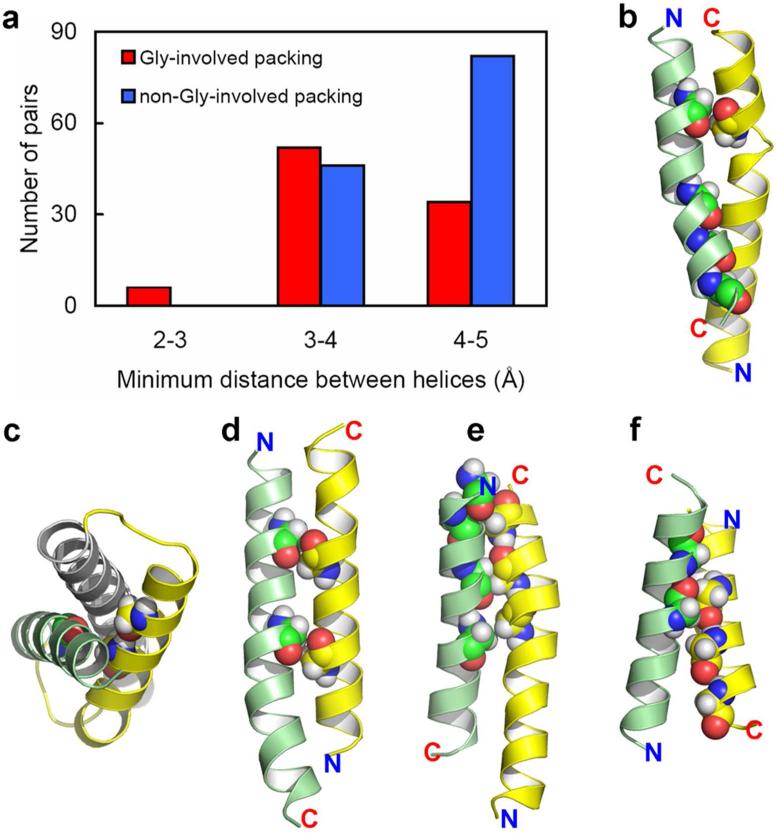
Among the growing number of membrane protein structures in the Protein Data Bank, there are many transmembrane domains that appear to be native-like; at the same time, there are others that appear to have less than complete native-like character. Hence, there is an increasing need for validation tools that distinguish native-like from non-native-like structures. Membrane mimetics used in protein structural characterizations differ in numerous physicochemical properties from native membranes and provide many opportunities for introducing non-native-like features into membrane protein structures. One possible approach for validating membrane protein structures is based on the use of glycine residues in transmembrane domains. Here, we have reviewed the membrane protein structure database and identified a set of benchmark proteins that appear to be native-like. In these structures, conserved glycine residues rarely face the lipid interstices, and many of them participate in close helix-helix packing. Glycine-based validation allowed the identification of non-native-like features in several membrane proteins and also shows the potential for verifying the native-like character for numerous other membrane protein structures.
Figures
References
-
- Tate CG. Comparison of three structures of the multidrug transporter EmrE. Curr Opin Struct Biol. 2006;16:457–464. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
