

Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis
- PMID: 22654667
- PMCID: PMC3360067
- DOI: 10.1371/journal.pgen.1002650
Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis
Abstract
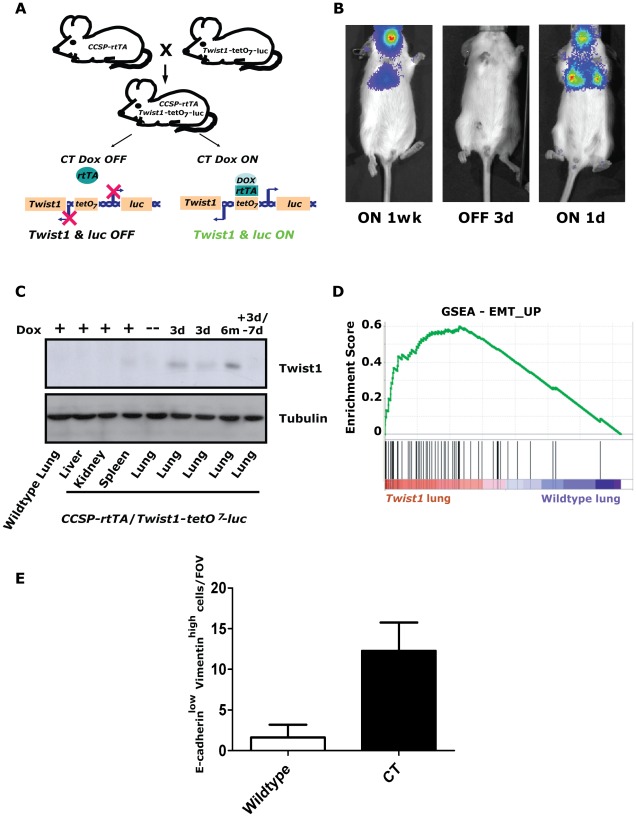
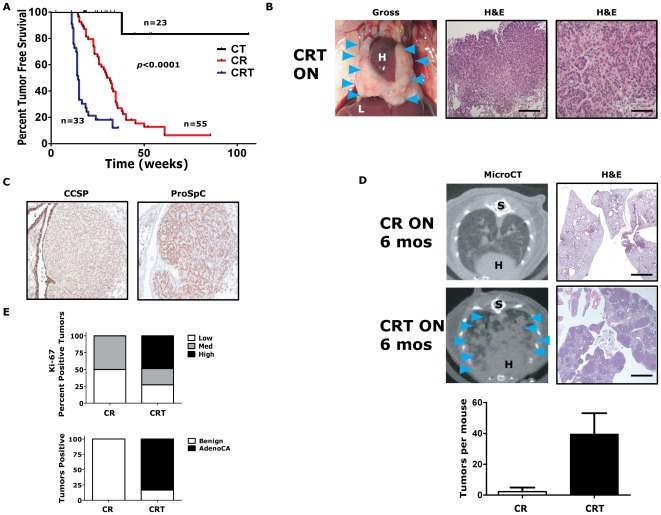
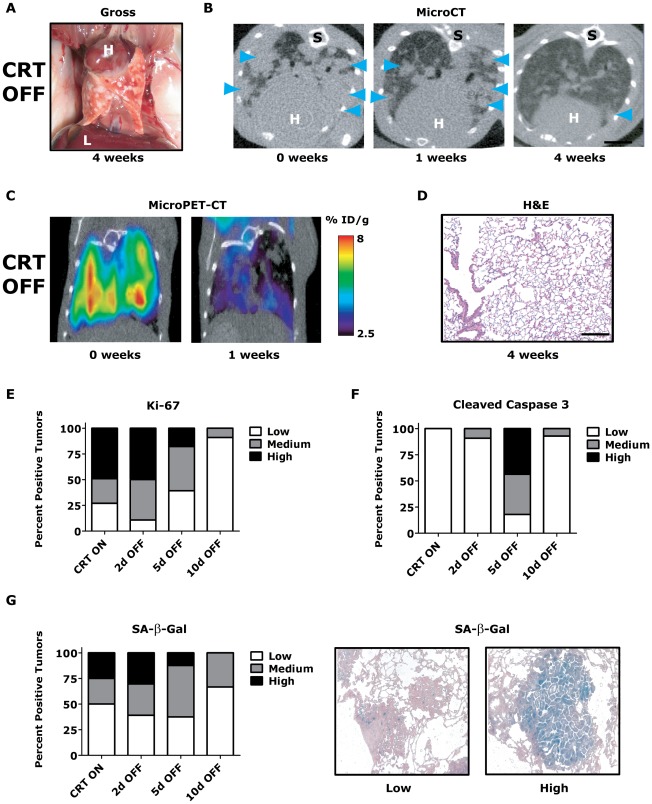
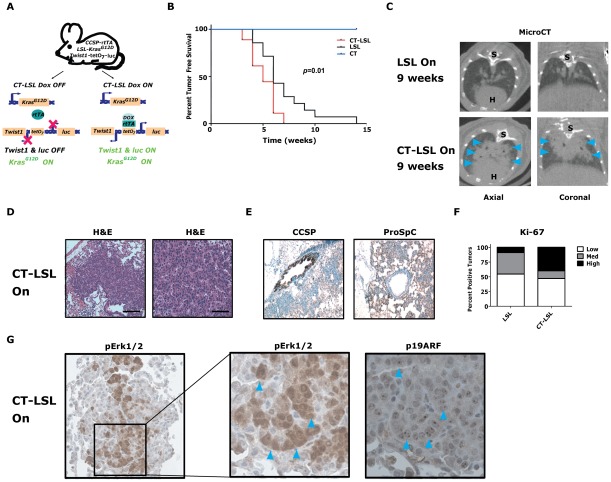
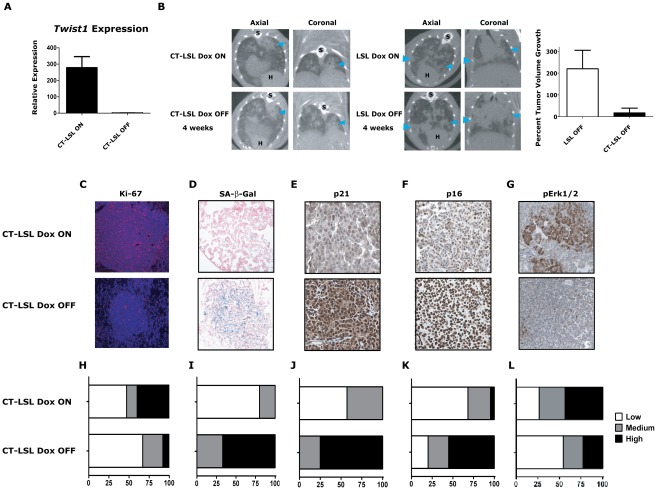
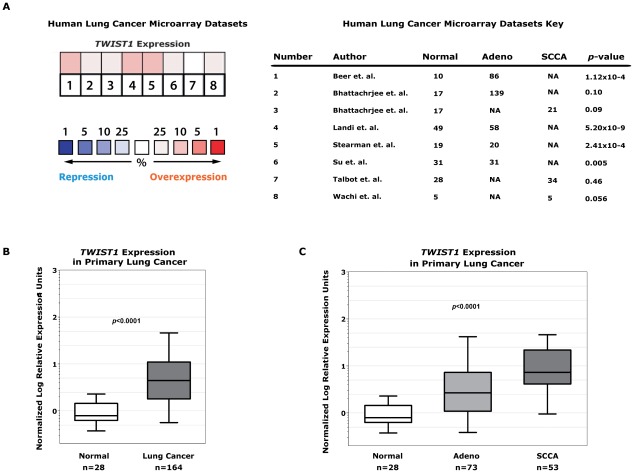
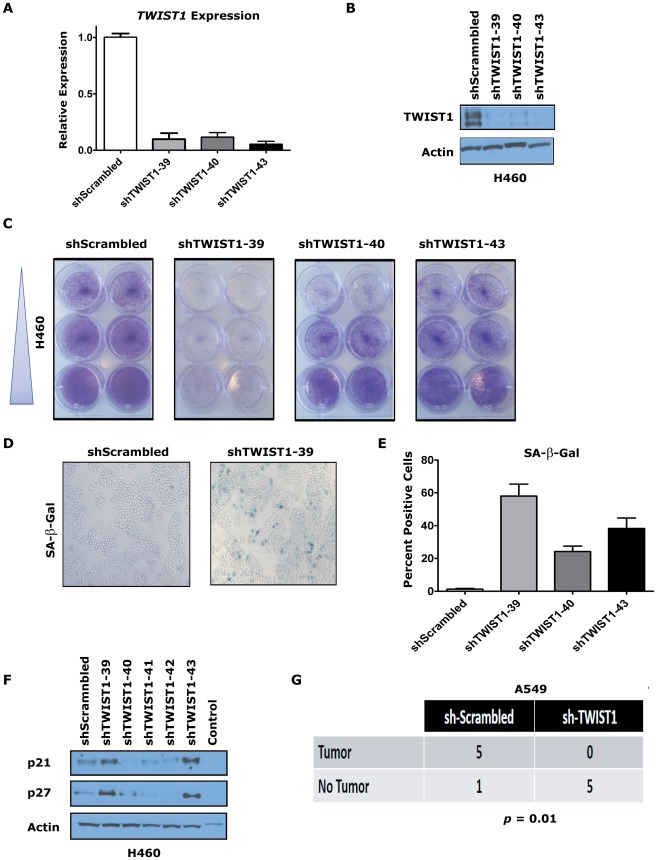
KRAS mutant lung cancers are generally refractory to chemotherapy as well targeted agents. To date, the identification of drugs to therapeutically inhibit K-RAS have been unsuccessful, suggesting that other approaches are required. We demonstrate in both a novel transgenic mutant Kras lung cancer mouse model and in human lung tumors that the inhibition of Twist1 restores a senescence program inducing the loss of a neoplastic phenotype. The Twist1 gene encodes for a transcription factor that is essential during embryogenesis. Twist1 has been suggested to play an important role during tumor progression. However, there is no in vivo evidence that Twist1 plays a role in autochthonous tumorigenesis. Through two novel transgenic mouse models, we show that Twist1 cooperates with Kras(G12D) to markedly accelerate lung tumorigenesis by abrogating cellular senescence programs and promoting the progression from benign adenomas to adenocarcinomas. Moreover, the suppression of Twist1 to physiological levels is sufficient to cause Kras mutant lung tumors to undergo senescence and lose their neoplastic features. Finally, we analyzed more than 500 human tumors to demonstrate that TWIST1 is frequently overexpressed in primary human lung tumors. The suppression of TWIST1 in human lung cancer cells also induced cellular senescence. Hence, TWIST1 is a critical regulator of cellular senescence programs, and the suppression of TWIST1 in human tumors may be an effective example of pro-senescence therapy.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. - PubMed
-
- Ji H, Li D, Chen L, Shimamura T, Kobayashi S, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
