

Alzheimer's disease: pathological mechanisms and recent insights
- PMID: 22654725
- PMCID: PMC3263461
- DOI: 10.2174/157015911798376181
Alzheimer's disease: pathological mechanisms and recent insights
Abstract
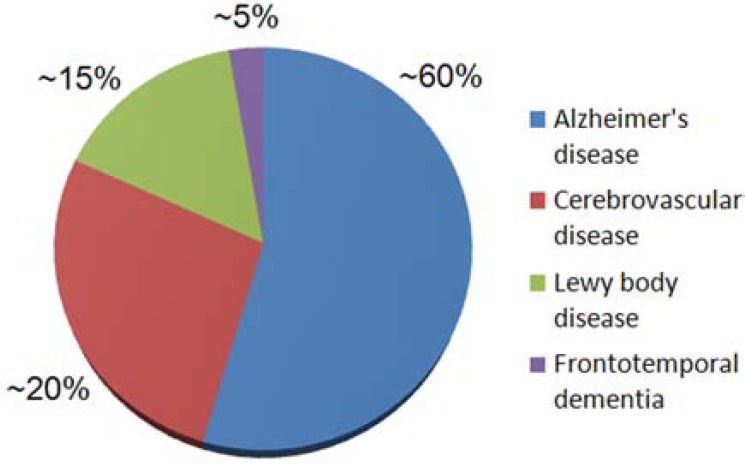
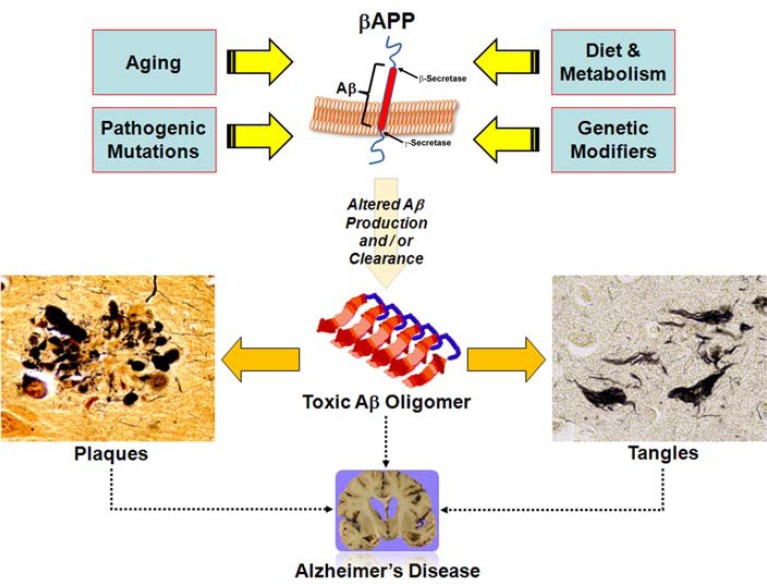
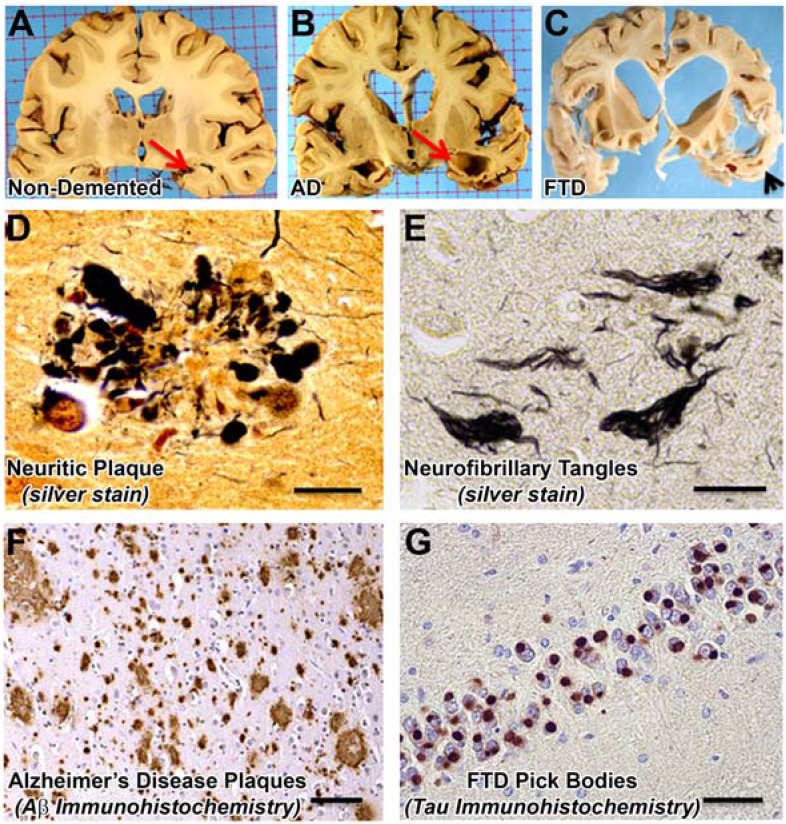
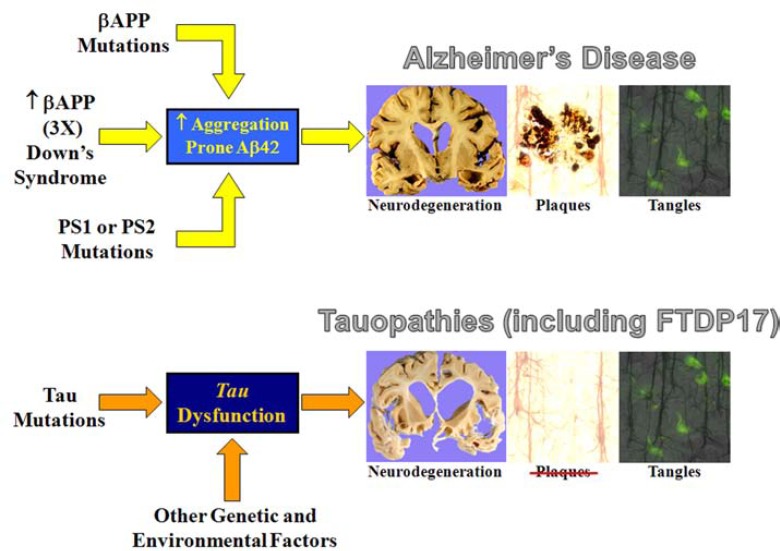
Amyloidopathies cause neurodegeneration in a substantial portion of the elderly population. Improvements in long term health care have made elderly individuals a large and growing demographic group, marking these diseases as a major public health concern. Alzheimer's Disease (AD) is the most studied form of neurodegenerative amyloidopathy. Although our understanding of AD is far from complete, several decades of research have advanced our knowledge to the point where it is conceivable that some form of disease modifying therapy may be available in the near future. These advances have been built on a strong mechanistic understanding of the disease from its underlying genetics, molecular biology and clinical pathology. Insights derived from the study of other neurodegenerative diseases, such as some forms of frontotemporal dementia, have been critical to this process. This knowledge has allowed researchers to construct animal models of the disease process that have paved the way towards the development of therapeutics. However, what was once thought to be a straightforward problem has evolved into a series of disappointing outcomes. Examination of pathways common to all neurodegenerative diseases, including the cellular mechanisms that clear misfolded proteins and their regulation, may be the best way to move forward.
Keywords: Amyloid; Tau.; amyloid-β peptide; amyloid-β precursor protein; neurodegeneration; protein misfolding; proteinopathies.
Figures
References
-
- He W, Sengupta M, Velkoff VA, DeBarros KA. 65+ in the United States. In: Commerce, U.S.D.o., Ed. Washington DC: US Government Printing Office; 2005. p. 254.
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. - PubMed
-
- Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984;122:1131–1135. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources