

Glutathione supplementation attenuates lipopolysaccharide-induced mitochondrial dysfunction and apoptosis in a mouse model of acute lung injury
- PMID: 22654772
- PMCID: PMC3361071
- DOI: 10.3389/fphys.2012.00161
Glutathione supplementation attenuates lipopolysaccharide-induced mitochondrial dysfunction and apoptosis in a mouse model of acute lung injury
Abstract
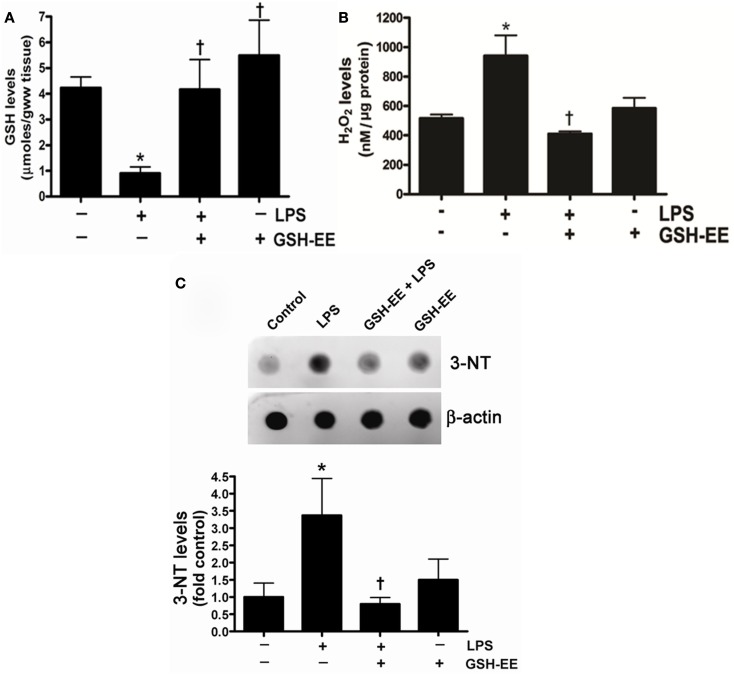
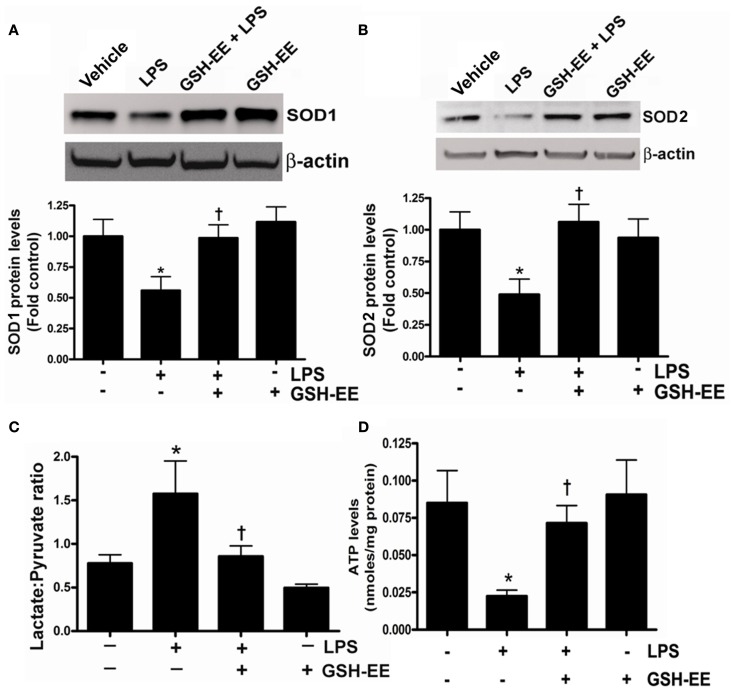
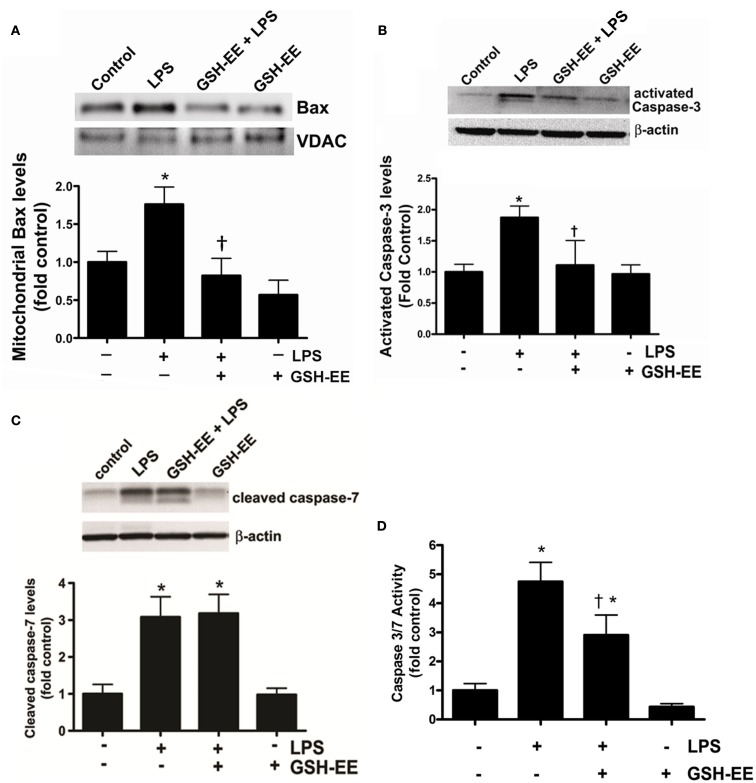
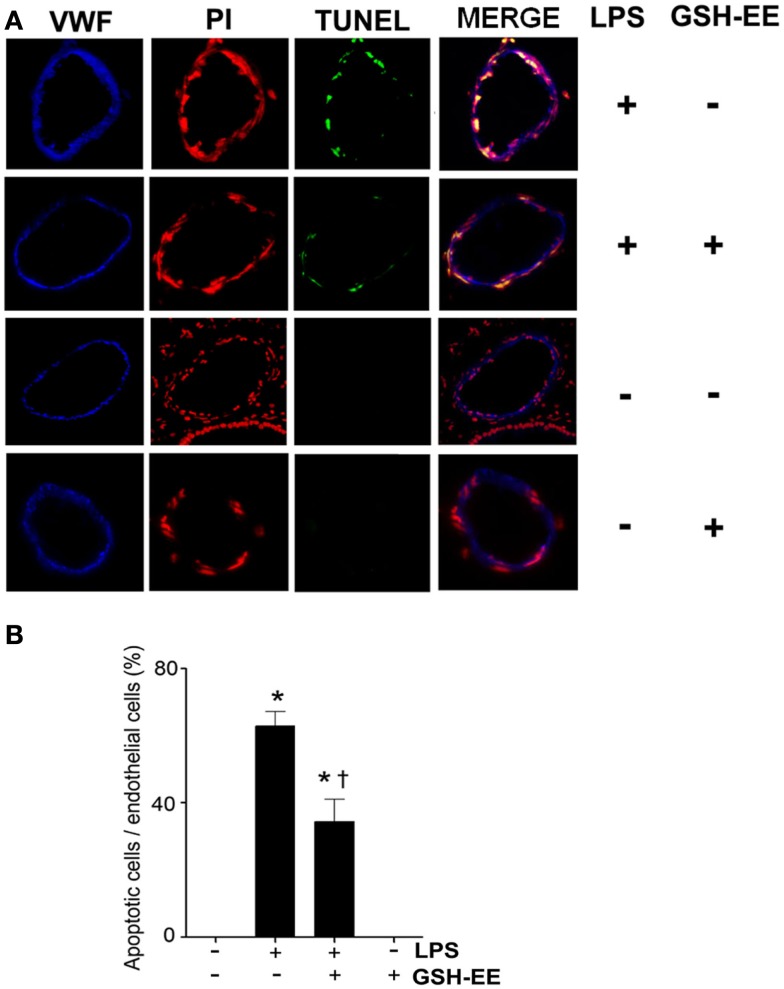
Acute lung injury (ALI) is a life threatening condition associated with hypoxemia, diffuse alveolar damage, inflammation, and loss of lung function. Lipopolysaccharide (LPS; endotoxin) from the outer membrane of Gram-negative bacteria is a major virulence factor involved in the development of ALI. The depletion of glutathione (GSH), an essential intra- and extra-cellular protective antioxidant, by LPS is an important event that contributes to the elevation in reactive oxygen species. Whether restoring GSH homeostasis can effectively ameliorate mitochondrial dysfunction and cellular apoptosis in ALI is unknown and therefore, was the focus of this study. In peripheral lung tissue of LPS-treated mice, hydrogen peroxide and protein nitration levels were significantly increased. Pre-treatment with GSH-ethyl ester (GSH-EE) prevented this increase in oxidative stress. LPS also increased the lactate/pyruvate ratio, attenuated SOD2 protein levels, and decreased ATP levels in the mouse lung indicative of mitochondrial dysfunction. Again, GSH-EE treatment preserved the mitochondrial function. Finally, our studies showed that LPS induced an increase in the mitochondrial translocation of Bax, caspase 3 activation, and nuclear DNA fragmentation and these parameters were all prevented with GSH-EE. Thus, this study suggests that GSH-EE supplementation may reduce the mitochondrial dysfunction associated with ALI.
Keywords: acute lung injury; apoptosis; glutathione ethyl ester; lipopolysaccharide; mitochondrial dysfunction.
Figures
References
-
- Behr J., Maier K., Degenkolb B., Krombach F., Vogelmeier C. (1997). Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am. J. Respir. Crit. Care Med. 156, 1897–1901 - PubMed
-
- Chuang C. Y., Chen T. L., Cherng Y. G., Tai Y. T., Chen T. G., Chen R. M. (2011). Lipopolysaccharide induces apoptotic insults to human alveolar epithelial A549 cells through reactive oxygen species-mediated activation of an intrinsic mitochondrion-dependent pathway. Arch. Toxicol. 85, 209–21810.1007/s00204-010-0585-x - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials