

Pathogenic Mechanisms and In Vitro Diagnosis of AERD
- PMID: 22654920
- PMCID: PMC3357963
- DOI: 10.1155/2012/789232
Pathogenic Mechanisms and In Vitro Diagnosis of AERD
Abstract
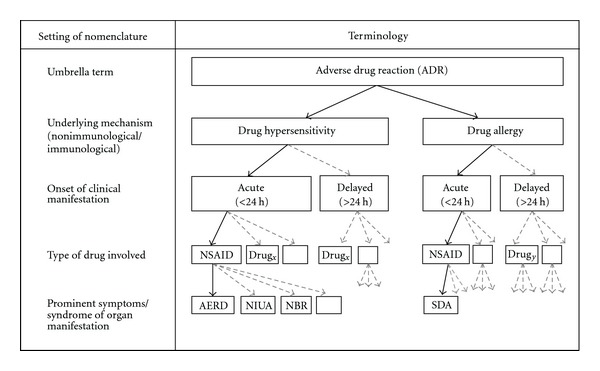
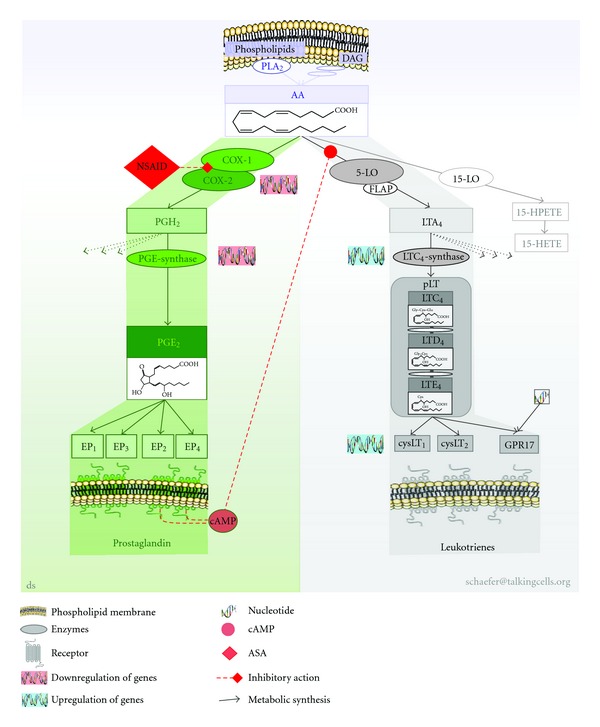
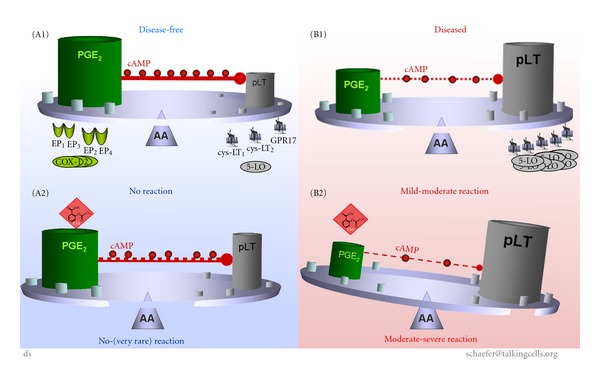
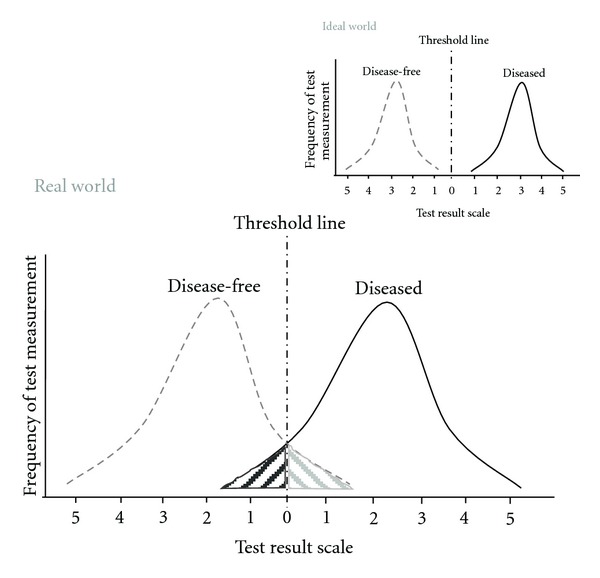
Aspirin-exacerbated respiratory disease (AERD) refers to chronic rhinosinusitis, nasal polyposis, bronchoconstriction, and/or eosinophilic inflammation in asthmatics following the exposure to nonsteroidal anti-inflammatory drugs (NSAIDs). A key pathogenic mechanism associated with AERD is the imbalance of eicosanoid metabolism focusing on prostanoid and leukotriene pathways in airway mucosa as well as blood cells. Genetic and functional metabolic studies on vital and non-vital cells pointed to the variability and the crucial role of lipid mediators in disease susceptibility and their response to medication. Eicosanoids, exemplified by prostaglandin E(2) (PGE(2)) and peptidoleukotrienes (pLT), are potential metabolic biomarkers contributing to the AERD phenotype. Also other mediators are implicated in the progress of AERD. Considering the various pathogenic mechanisms of AERD, a multitude of metabolic and genetic markers is suggested to be implicated and were introduced as potential biomarkers for in vitro diagnosis during the past decades. Deduced from an eicosanoid-related pathogenic mechanism, functional tests balancing PGE(2) and pLT as well as other eicosanoids from preferentially vital leukocytes demonstrated their applicability for in vitro diagnosis of AERD.
Figures
Similar articles
-
Aspirin or other nonsteroidal inflammatory agent exacerbated asthma.J Allergy Clin Immunol Pract. 2014 Nov-Dec;2(6):653-7. doi: 10.1016/j.jaip.2014.09.009. Epub 2014 Nov 6. J Allergy Clin Immunol Pract. 2014. PMID: 25439353 Review.
-
Genetic mechanisms in aspirin-exacerbated respiratory disease.J Allergy (Cairo). 2012;2012:794890. doi: 10.1155/2012/794890. Epub 2011 Aug 7. J Allergy (Cairo). 2012. PMID: 21837245 Free PMC article.
-
Mepolizumab targets multiple immune cells in aspirin-exacerbated respiratory disease.J Allergy Clin Immunol. 2021 Aug;148(2):574-584. doi: 10.1016/j.jaci.2021.05.043. Epub 2021 Jun 16. J Allergy Clin Immunol. 2021. PMID: 34144111 Free PMC article. Clinical Trial.
-
Update on recent advances in the management of aspirin exacerbated respiratory disease.Yonsei Med J. 2009 Dec 31;50(6):744-50. doi: 10.3349/ymj.2009.50.6.744. Epub 2009 Dec 18. Yonsei Med J. 2009. PMID: 20046412 Free PMC article. Review.
-
Inflammatory heterogeneity in aspirin-exacerbated respiratory disease.J Allergy Clin Immunol. 2021 Apr;147(4):1318-1328.e5. doi: 10.1016/j.jaci.2020.11.001. Epub 2020 Nov 12. J Allergy Clin Immunol. 2021. PMID: 33189729 Free PMC article.
Cited by
-
Emerging targets in lipid-based therapy.Biochem Pharmacol. 2013 Mar 1;85(5):673-688. doi: 10.1016/j.bcp.2012.11.028. Epub 2012 Dec 20. Biochem Pharmacol. 2013. PMID: 23261527 Free PMC article. Review.
-
Development of a genetic marker set to diagnose aspirin-exacerbated respiratory disease in a genome-wide association study.Pharmacogenomics J. 2015 Aug;15(4):316-21. doi: 10.1038/tpj.2014.78. Epub 2015 Feb 24. Pharmacogenomics J. 2015. PMID: 25707394
-
Aspirin-exacerbated respiratory disease and current treatment modalities.Eur Arch Otorhinolaryngol. 2017 Mar;274(3):1291-1300. doi: 10.1007/s00405-016-4273-1. Epub 2016 Aug 18. Eur Arch Otorhinolaryngol. 2017. PMID: 27538737 Review.
-
Aspirin sensitivity and chronic rhinosinusitis with polyps: a fatal combination.J Allergy (Cairo). 2012;2012:817910. doi: 10.1155/2012/817910. Epub 2012 Aug 14. J Allergy (Cairo). 2012. PMID: 22927869 Free PMC article.
-
Integrative information theoretic network analysis for genome-wide association study of aspirin exacerbated respiratory disease in Korean population.BMC Med Genomics. 2017 May 24;10(Suppl 1):31. doi: 10.1186/s12920-017-0266-1. BMC Med Genomics. 2017. PMID: 28589859 Free PMC article.
References
-
- Hirschberg. Mitteilungen über einen Fall von Nebenwirkungen des Aspirin. Dt Med Wochenschr. 1902;28:416–417.
-
- Widal F, Abrami P, Lermoyez J. Anaphylaxie et idiosyncrasie. La Presse Medicale. 1922;30:189–193.
-
- Samter M, Beers RF. Concerning the nature of intolerance to aspirin. Journal of Allergy. 1967;40(5):281–293. - PubMed
-
- Delaney JC. The diagnosis of aspirin idiosyncrasy by analgesic challenge. Clinical Allergy. 1976;6(2):177–181. - PubMed
-
- Stevenson DD, Zuraw BL. Pathogenesis of aspirin-exacerbated respiratory disease. Clinical Reviews in Allergy and Immunology. 2003;24(2):169–187. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous