

Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress
- PMID: 22657349
- PMCID: PMC3887453
- DOI: 10.1089/ars.2012.4609
Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress
Abstract
Significance: Oxidative stress is a well established hallmark of cardiovascular disease and there is strong evidence for a causal role of reactive oxygen and nitrogen species (RONS) therein.
Recent advances: Improvement of cardiovascular complications by genetic deletion of RONS producing enzymes and overexpression of RONS degrading enzymes proved the involvement of these species in cardiovascular disease at a molecular level. Vice versa, overexpression of RONS producing enzymes as well as deletion of antioxidant enzymes was demonstrated to aggravate cardiovascular complications.
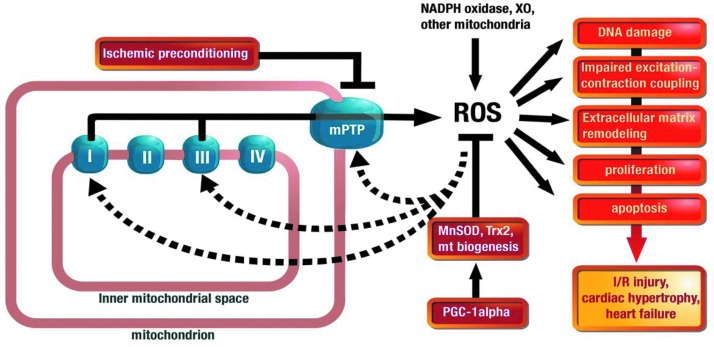
Critical issues: With the present overview we present and discuss different pathways how mitochondrial RONS interact (crosstalk) with other sources of oxidative stress, namely NADPH oxidases, xanthine oxidase and an uncoupled nitric oxide synthase. The potential mechanisms of how this crosstalk proceeds are discussed in detail. Several examples from the literature are summarized (including hypoxia, angiotensin II mediated vascular dysfunction, cellular starvation, nitrate tolerance, aging, hyperglycemia, β-amyloid stress and others) and the underlying mechanisms are put together to a more general concept of redox-based activation of different sources of RONS via enzyme-specific "redox switches". Mitochondria play a key role in this concept providing redox triggers for oxidative damage in the cardiovascular system but also act as amplifiers to increase the burden of oxidative stress.
Future directions: Based on these considerations, the characterization of the role of mitochondrial RONS formation in cardiac disease as well as inflammatory processes but also the role of mitochondria as potential therapeutic targets in these pathophysiological states should be addressed in more detail in the future.
Figures






References
-
- Abbas AK. and Lichtman AH. Basic Immunology: Functions and Disorders of the Immune System. Philadelphia, PA: Saunders/Elsevier; 2009, pp. viii, 312
-
- AbdAlla S, Lother H, Langer A, el Faramawy Y, and Quitterer U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 119: 343–354, 2004 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
