

Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene
- PMID: 22661473
- PMCID: PMC3394687
- DOI: 10.1167/iovs.11-9166
Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene
Abstract
Purpose: We evaluated the pathogenicity of the G1961E mutation in the ABCA4 gene, and present the range of retinal phenotypes associated with this mutation in homozygosity in a patient cohort with ABCA4-associated phenotypes.
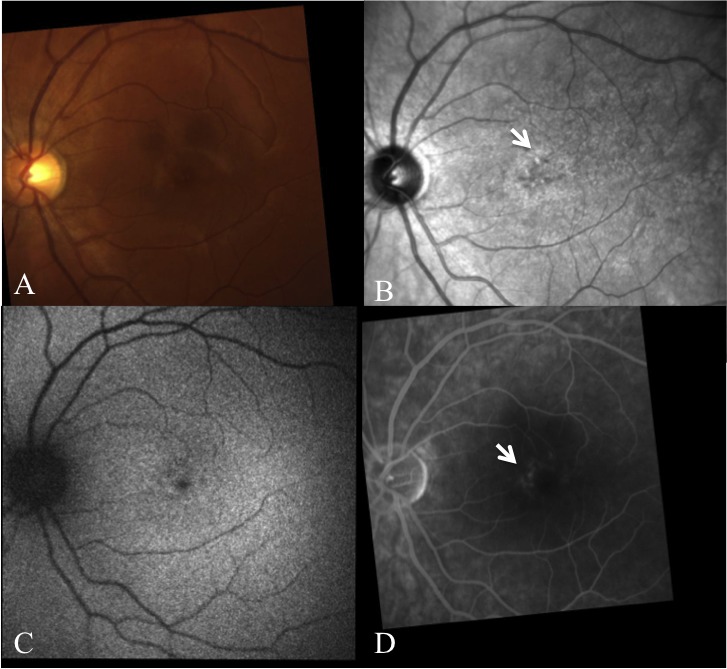
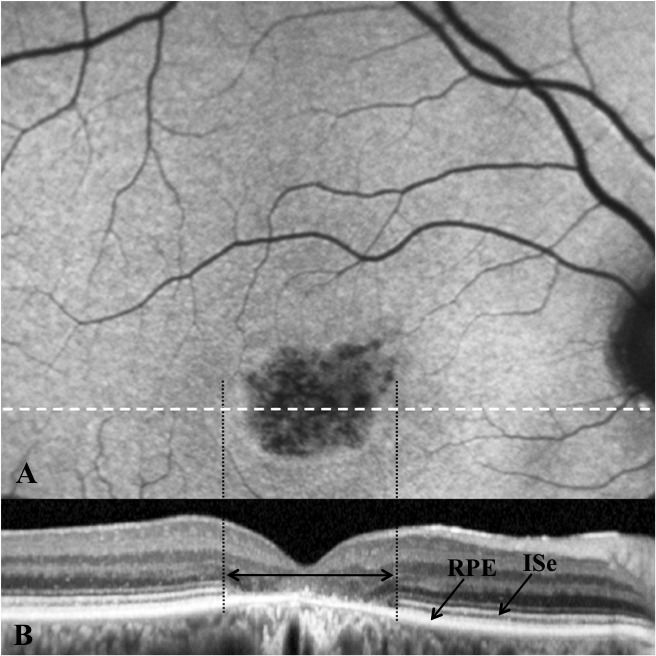
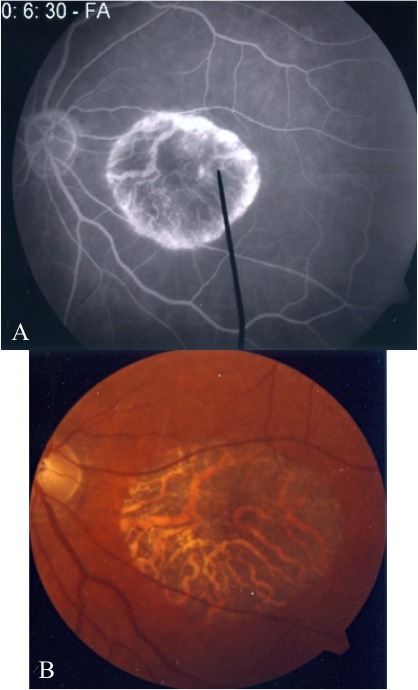
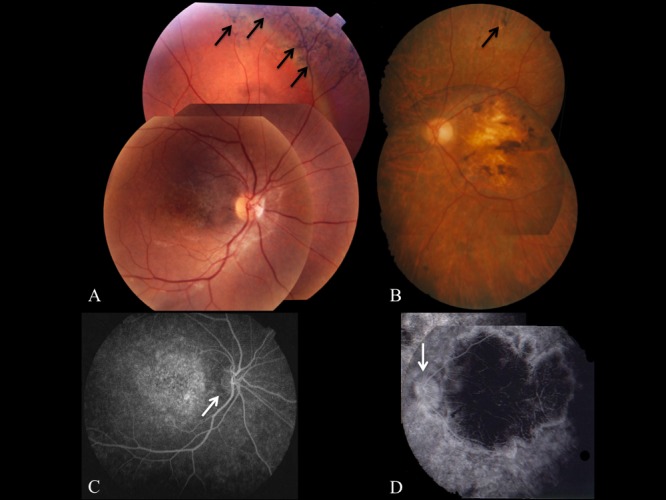
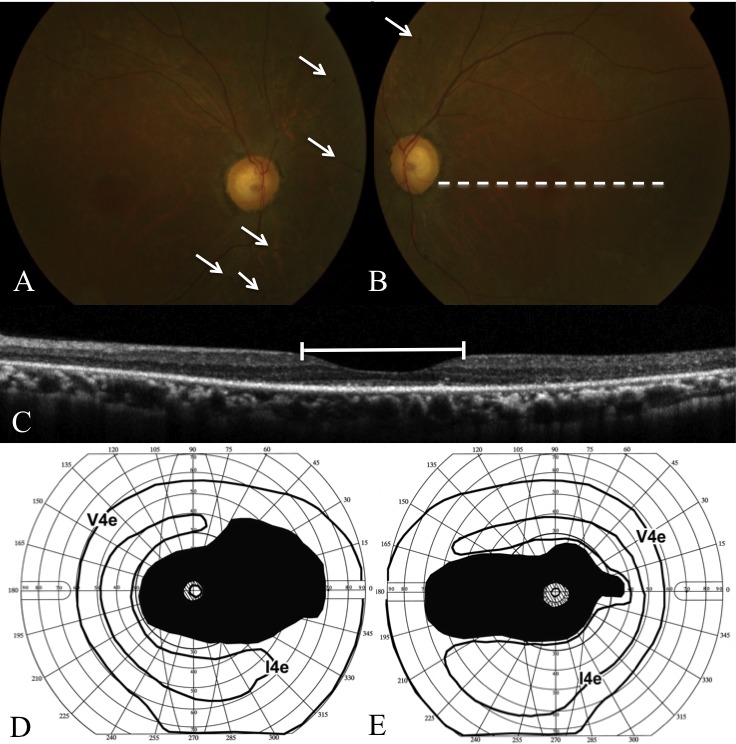
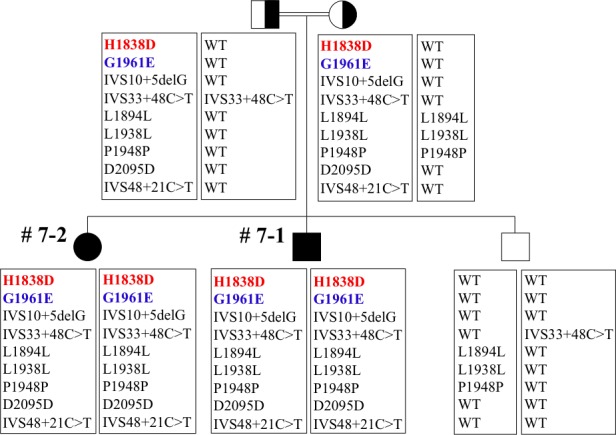
Methods: Patients were enrolled from the ABCA4 disease database at Columbia University or by inquiry from collaborating physicians. Only patients homozygous for the G1961E mutation were enrolled. The entire ABCA4 gene open reading frame, including all exons and flanking intronic sequences, was sequenced in all patients. Phenotype data were obtained from clinical history and examination, fundus photography, infrared imaging, fundus autofluorescence, fluorescein angiography, and spectral domain-optical coherence tomography. Additional functional data were obtained using the full-field electroretinogram, and static or kinetic perimetry.
Results: We evaluated 12 patients homozygous for the G1961E mutation. All patients had evidence of retinal pathology consistent with the range of phenotypes observed in ABCA4 disease. The latest age of onset was recorded at 64 years, in a patient diagnosed initially with age-related macular degeneration (AMD). Of 6 patients in whom severe structural (with/without functional) fundus changes were detected, 5 had additional, heterozygous or homozygous, variants detected in the ABCA4 gene.
Conclusions: Homozygous G1961E mutation in ABCA4 results in a range of retinal pathology. The phenotype usually is at the milder end of the disease spectrum, with severe phenotypes linked to the presence of additional ABCA4 variants. Our report also highlights that milder, late-onset Stargardt disease may be confused with AMD.
Conflict of interest statement
Disclosure:
Figures
Comment in
-
Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene.Invest Ophthalmol Vis Sci. 2013 Jan 17;54(1):520. doi: 10.1167/iovs.12-11472. Invest Ophthalmol Vis Sci. 2013. PMID: 23329737 No abstract available.
-
Author response: Retinal phenotypes in patients homozygous for the G1961E mutation in the ABCA4 gene.Invest Ophthalmol Vis Sci. 2013 Jan 17;54(1):521. doi: 10.1167/iovs.12-11528. Invest Ophthalmol Vis Sci. 2013. PMID: 23329738 No abstract available.
References
-
- Blacharski P. Fundus flavimaculatus. : Newsome DA, Retinal Dystrophies and Degenerations. New York, NY: Raven Press; 1988:382
-
- Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature Genetics. 1997;15:236–246 - PubMed
-
- Jaakson K, Zernant J, Külm M, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403 - PubMed
-
- Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR. Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet. 2001;108:346–355 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
