

Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms
- PMID: 22661952
- PMCID: PMC3357552
- DOI: 10.3389/fphys.2012.00138
Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms
Abstract
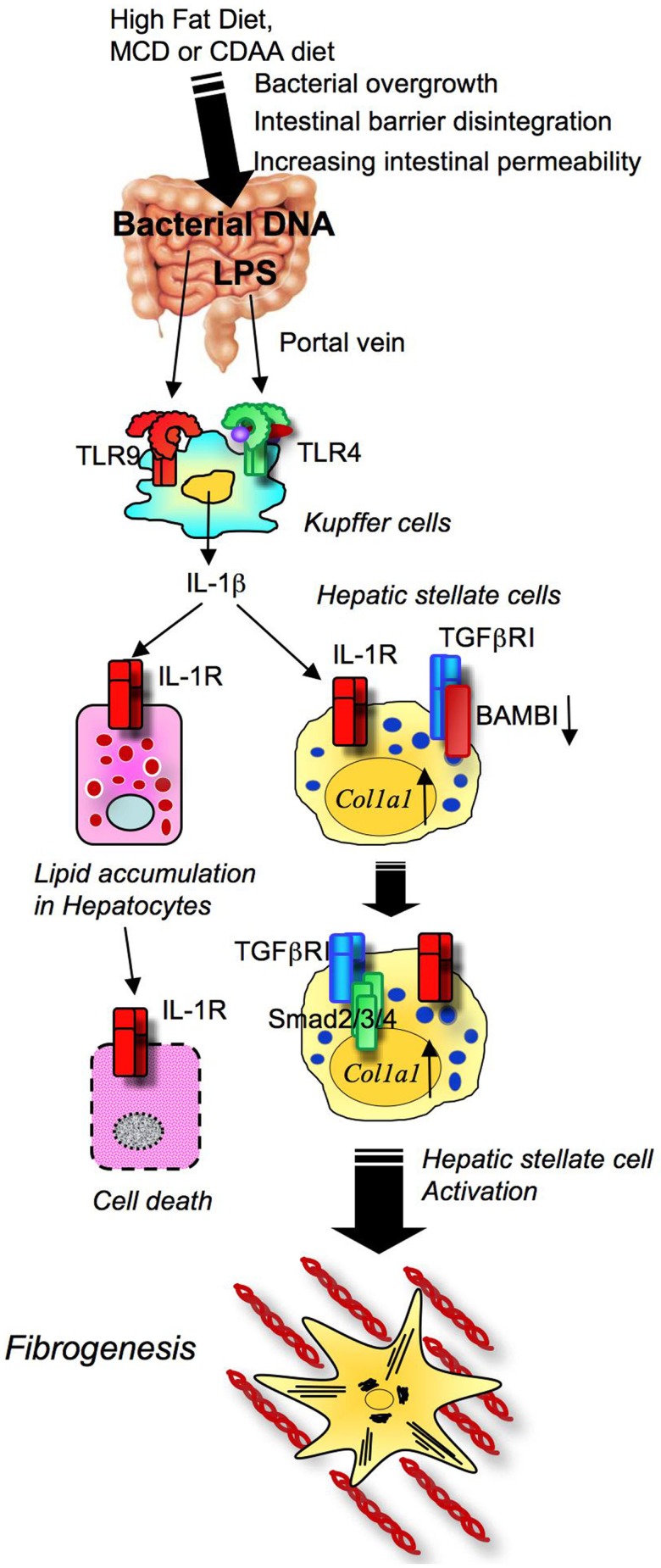
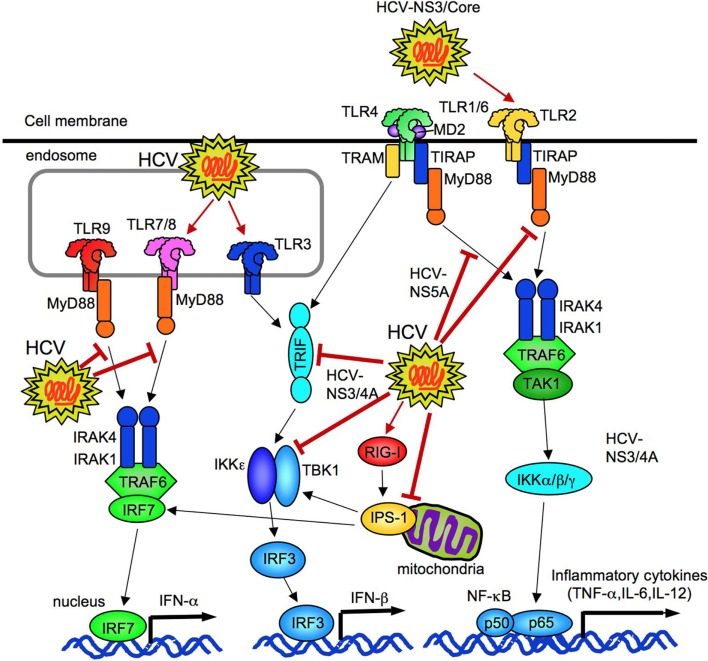
Toll-like receptors (TLRs) are pattern recognition receptors that distinguish conserved microbial products, also known as pathogen-associated molecular patterns (PAMPs), from host molecules. Liver is the first filter organ between the gastrointestinal tracts and the rest of the body through portal circulation. Thus, the liver is a major organ that must deal with PAMPs and microorganisms translocated from the intestine and to respond to the damage associated molecular patterns (DAMPs) released from injured organs. These PAMPs and DAMPs preferentially activate TLR signaling on various cell types in the liver inducing the production of inflammatory and fibrogenic cytokines that initiate and prolong liver inflammation, thereby leading to fibrosis. We summarize recent findings on the role of TLRs, ligands, and intracellular signaling in the pathophysiology of liver fibrosis due to different etiology, as well as to highlight the potential role of TLR signaling in liver fibrosis associated with hepatitis C infection, non-alcoholic and alcoholic steatoheoatitis, primary biliary cirrhosis, and cystic fibrosis.
Keywords: alcoholic liver disease; cystic fibrosis; hepatitis C; non-alcoholic steatohepatitis; primary biliary cirrhosis; toll-like receptors.
Figures
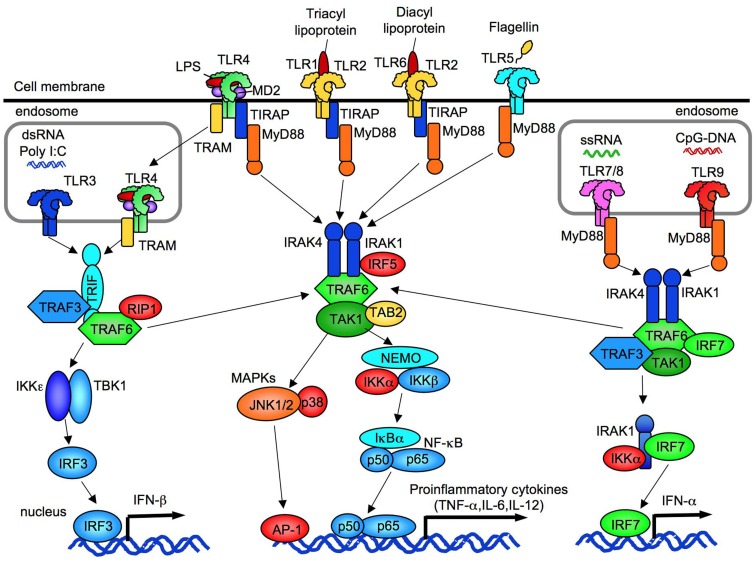
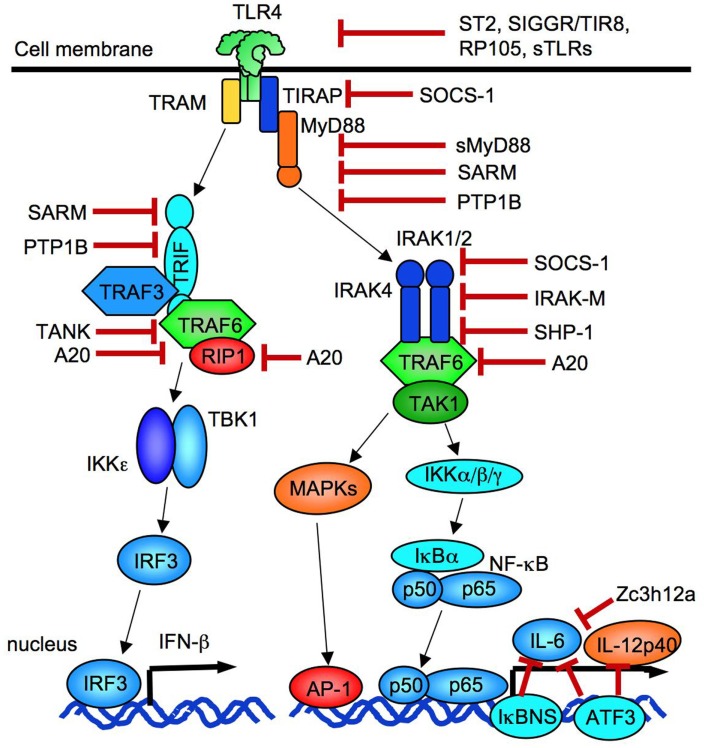
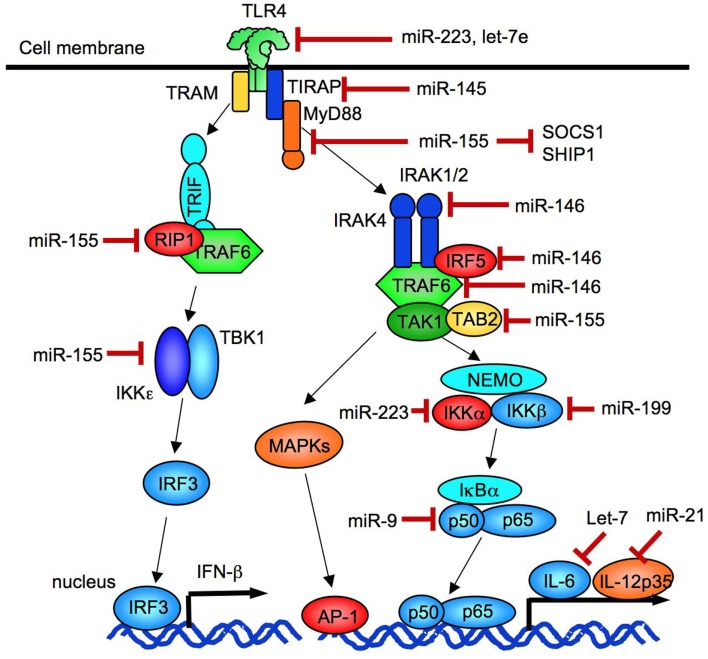

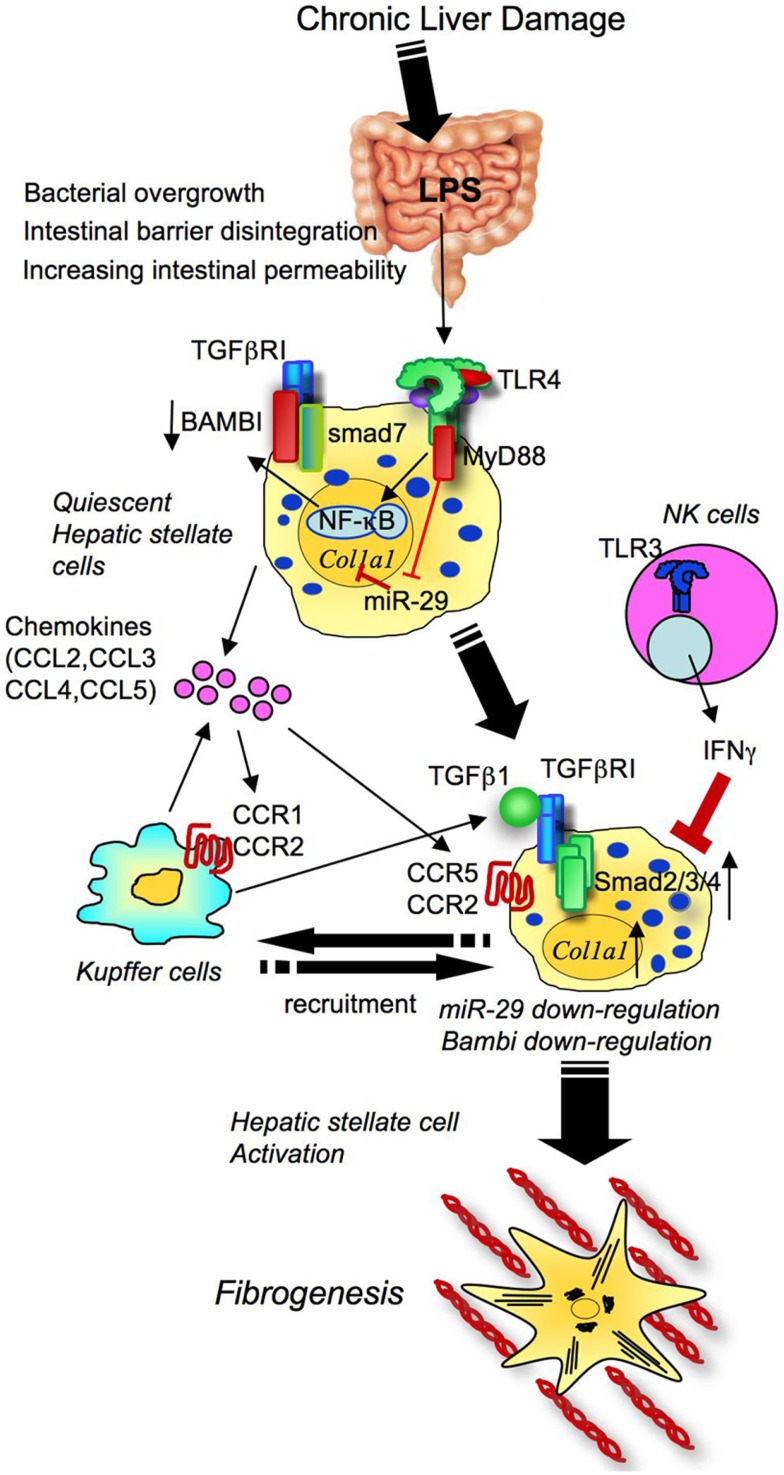

References
-
- Abe T., Kaname Y., Hamamoto I., Tsuda Y., Wen X., Taguwa S., Moriishi K., Takeuchi O., Kawai T., Kanto T., Hayashi N., Akira S., Matsuura Y. (2007). Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 81, 8953–896610.1128/JVI.00649-07 - DOI - PMC - PubMed
-
- Ambrosini Y. M., Yang G. X., Zhang W., Tsuda M., Shu S., Tsuneyama K., Leung P. S., Ansari A. A., Coppel R. L., Gershwin M. E. (2011). The multi-hit hypothesis of primary biliary cirrhosis: polyinosinic-polycytidylic acid (poly I:C) and murine autoimmune cholangitis. Clin. Exp. Immunol. 166, 110–12010.1111/j.1365-2249.2011.04453.x - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources