

Regulation of RKIP function by Helicobacter pylori in gastric cancer
- PMID: 22662230
- PMCID: PMC3360604
- DOI: 10.1371/journal.pone.0037819
Regulation of RKIP function by Helicobacter pylori in gastric cancer
Abstract
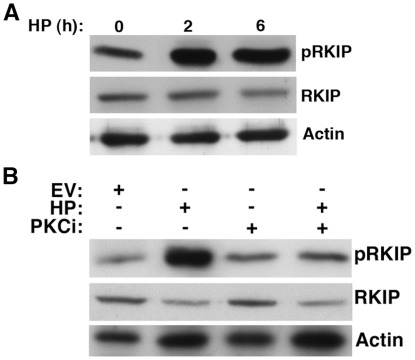
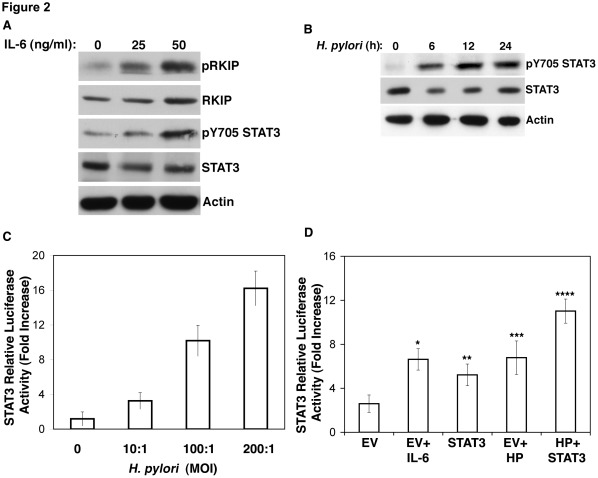
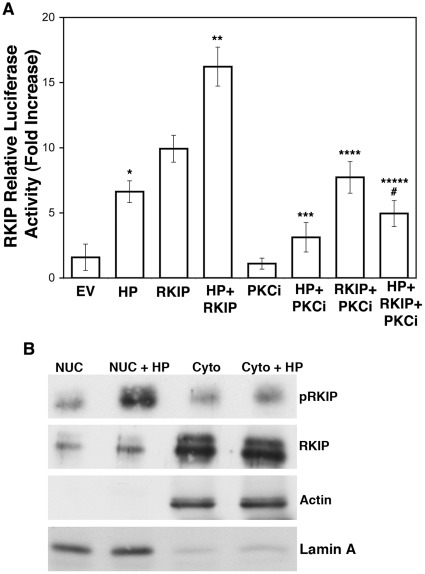
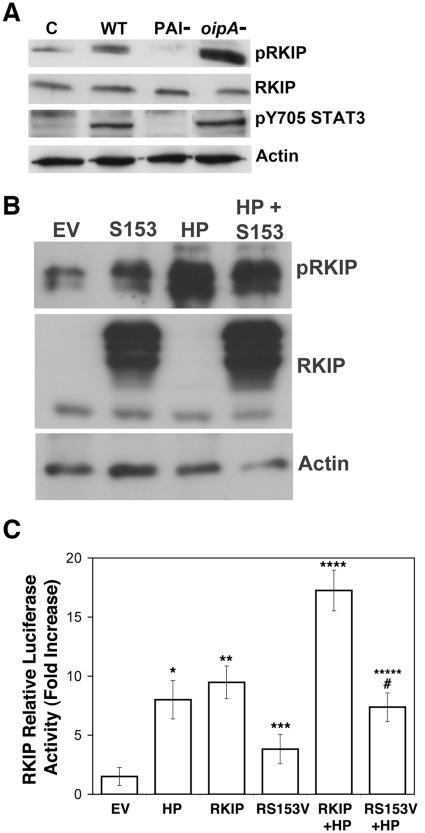
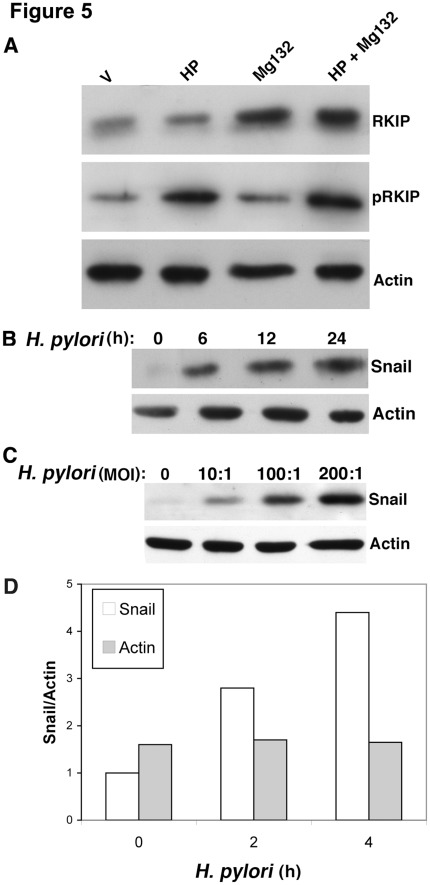
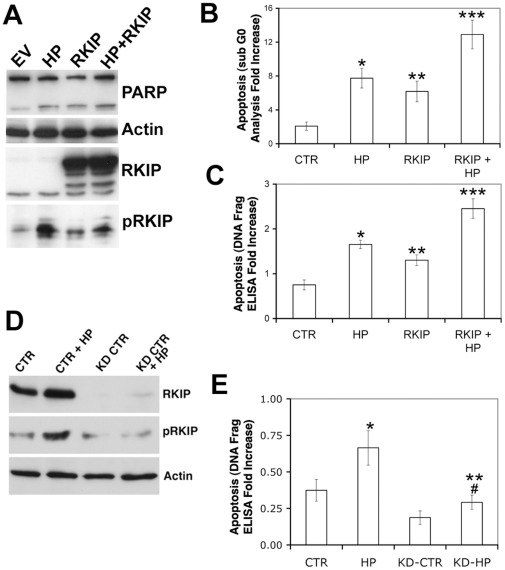
Helicobacter pylori (H. pylori) is a gram-negative, spiral-shaped bacterium that infects more than half of the world's population and is a major cause of gastric adenocarcinoma. The mechanisms that link H. pylori infection to gastric carcinogenesis are not well understood. In the present study, we report that the Raf-kinase inhibitor protein (RKIP) has a role in the induction of apoptosis by H. pylori in gastric epithelial cells. Western blot and luciferase transcription reporter assays demonstrate that the pathogenicity island of H. pylori rapidly phosphorylates RKIP, which then localizes to the nucleus where it activates its own transcription and induces apoptosis. Forced overexpression of RKIP enhances apoptosis in H. pylori-infected cells, whereas RKIP RNA inhibition suppresses the induction of apoptosis by H. pylori infection. While inducing the phosphorylation of RKIP, H. pylori simultaneously targets non-phosphorylated RKIP for proteasome-mediated degradation. The increase in RKIP transcription and phosphorylation is abrogated by mutating RKIP serine 153 to valine, demonstrating that regulation of RKIP activity by H. pylori is dependent upon RKIP's S153 residue. In addition, H. pylori infection increases the expression of Snail, a transcriptional repressor of RKIP. Our results suggest that H. pylori utilizes a tumor suppressor protein, RKIP, to promote apoptosis in gastric cancer cells.
Conflict of interest statement
Figures
References
-
- Garcia M, Jemal A, Ward E Center M, Hao Y, et al. American Cancer Society; 2007. Global Cancer Facts & Figures 2007. pp. 1–52.
-
- Wang TC, Goldenring JR, Dangler C, Ito S, Mueller A, et al. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology. 1998;114:675–689. - PubMed
-
- Brenes F, Ruiz B, Correa P, Hunter F, Rhamakrishnan T, et al. Helicobacter pylori causes hyperproliferation of the gastric epithelium: pre- and post-eradication indices of proliferating cell nuclear antigen. Am J Gastroenterol. 1993;88:1870–1875. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
