

Induction of neuronal death by microglial AGE-albumin: implications for Alzheimer's disease
- PMID: 22662249
- PMCID: PMC3360664
- DOI: 10.1371/journal.pone.0037917
Induction of neuronal death by microglial AGE-albumin: implications for Alzheimer's disease
Abstract
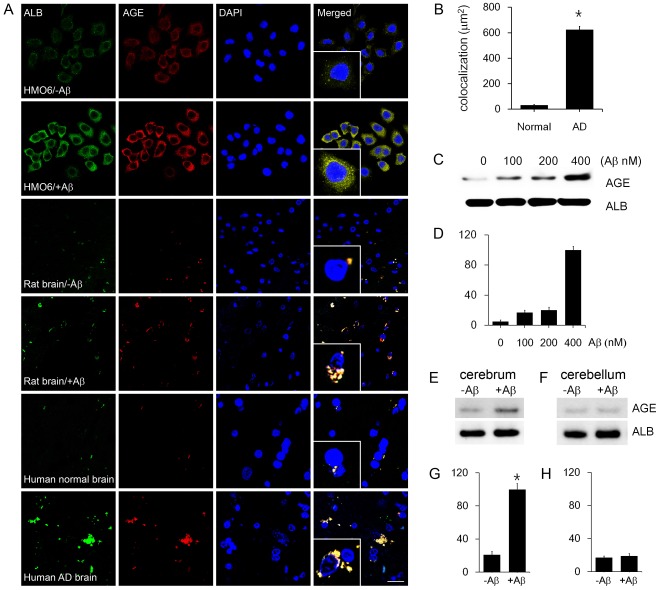
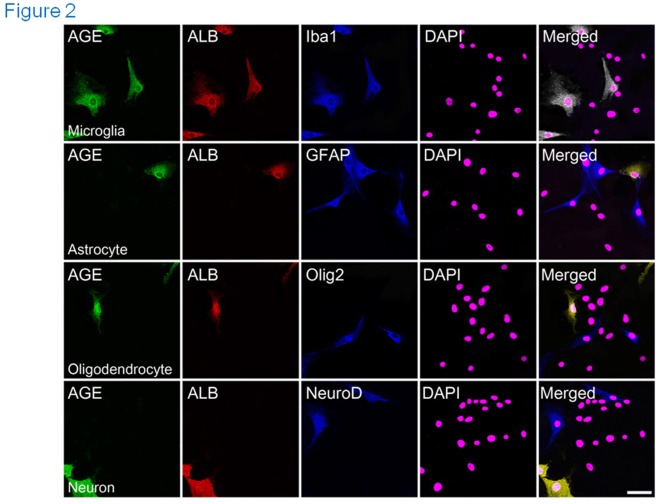
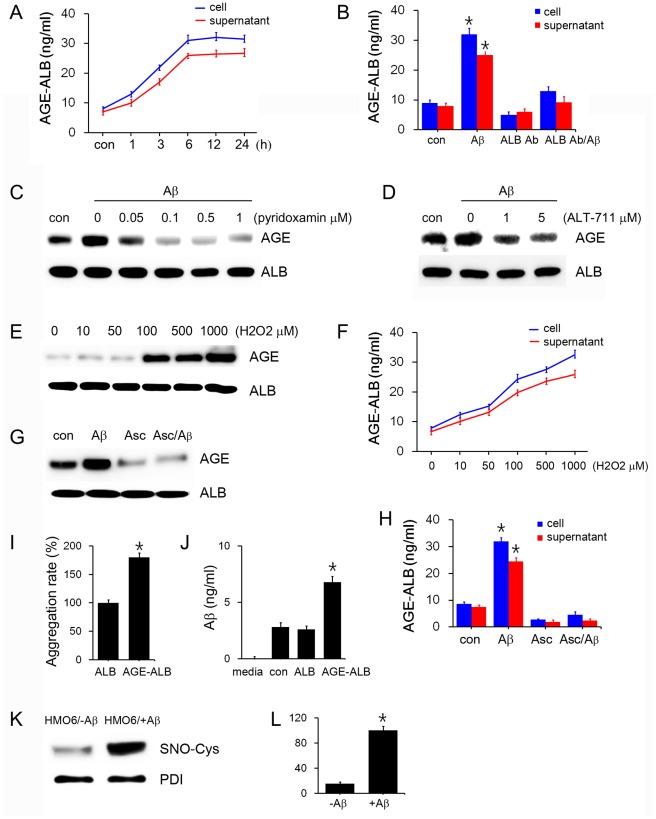
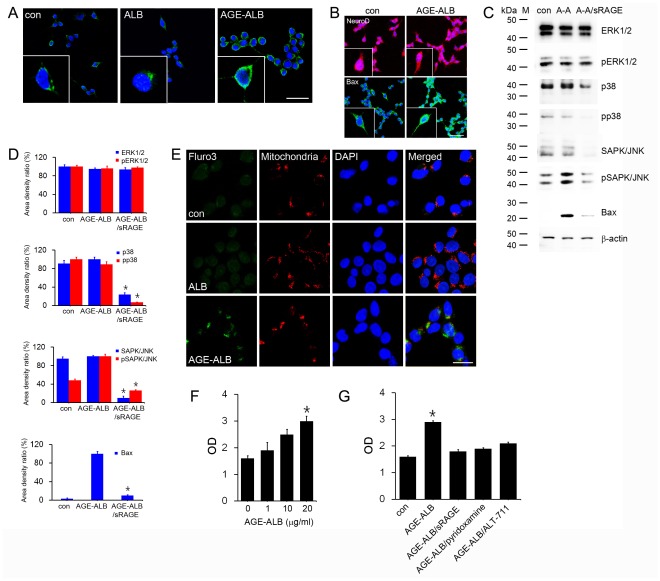
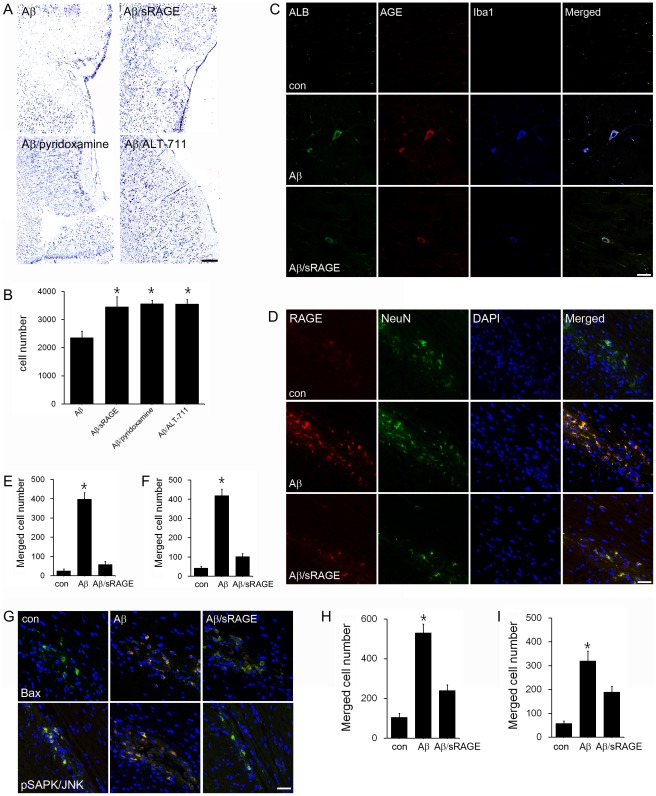
Advanced glycation end products (AGEs) have long been considered as potent molecules promoting neuronal cell death and contributing to neurodegenerative disorders such as Alzheimer's disease (AD). In this study, we demonstrate that AGE-albumin, the most abundant AGE product in human AD brains, is synthesized in activated microglial cells and secreted into the extracellular space. The rate of AGE-albumin synthesis in human microglial cells is markedly increased by amyloid-β exposure and oxidative stress. Exogenous AGE-albumin upregulates the receptor protein for AGE (RAGE) and augments calcium influx, leading to apoptosis of human primary neurons. In animal experiments, soluble RAGE (sRAGE), pyridoxamine or ALT-711 prevented Aβ-induced neuronal death in rat brains. Collectively, these results provide evidence for a new mechanism by which microglial cells promote death of neuronal cells through synthesis and secretion of AGE-albumin, thereby likely contributing to neurodegenerative diseases such as AD.
Conflict of interest statement
Figures
Similar articles
-
Preventing activation of receptor for advanced glycation endproducts in Alzheimer's disease.Curr Drug Targets CNS Neurol Disord. 2005 Jun;4(3):249-66. doi: 10.2174/1568007054038210. Curr Drug Targets CNS Neurol Disord. 2005. PMID: 15975028 Review.
-
Protection against RAGE-mediated neuronal cell death by sRAGE-secreting human mesenchymal stem cells in 5xFAD transgenic mouse model.Brain Behav Immun. 2017 Nov;66:347-358. doi: 10.1016/j.bbi.2017.07.158. Epub 2017 Jul 29. Brain Behav Immun. 2017. PMID: 28760504
-
Microglial AGE-albumin is critical for neuronal death in Parkinson's disease: a possible implication for theranostics.Int J Nanomedicine. 2016 Aug 23;10 Spec Iss(Spec Iss):281-92. doi: 10.2147/IJN.S95077. eCollection 2015. Int J Nanomedicine. 2016. PMID: 27601894 Free PMC article.
-
Site-specific blockade of RAGE-Vd prevents amyloid-beta oligomer neurotoxicity.J Neurosci. 2008 May 14;28(20):5149-58. doi: 10.1523/JNEUROSCI.4878-07.2008. J Neurosci. 2008. PMID: 18480271 Free PMC article.
-
AGE-RAGE stress: a changing landscape in pathology and treatment of Alzheimer's disease.Mol Cell Biochem. 2019 Sep;459(1-2):95-112. doi: 10.1007/s11010-019-03553-4. Epub 2019 May 11. Mol Cell Biochem. 2019. PMID: 31079281 Review.
Cited by
-
Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress.Brain Res. 2016 Apr 15;1637:34-55. doi: 10.1016/j.brainres.2016.02.016. Epub 2016 Feb 13. Brain Res. 2016. PMID: 26883165 Free PMC article. Review.
-
Glycation exacerbates the neuronal toxicity of β-amyloid.Cell Death Dis. 2013 Jun 13;4(6):e673. doi: 10.1038/cddis.2013.180. Cell Death Dis. 2013. PMID: 23764854 Free PMC article.
-
Newer Therapeutic Approaches in Treating Alzheimer's Disease: A Comprehensive Review.ACS Omega. 2025 Feb 3;10(6):5148-5171. doi: 10.1021/acsomega.4c05527. eCollection 2025 Feb 18. ACS Omega. 2025. PMID: 39989768 Free PMC article. Review.
-
Mechanism and implications of advanced glycation end products (AGE) and its receptor RAGE axis as crucial mediators linking inflammation and obesity.Mol Biol Rep. 2025 Jun 5;52(1):556. doi: 10.1007/s11033-025-10632-x. Mol Biol Rep. 2025. PMID: 40474023 Review.
-
CRISPR/Cas9 Edited sRAGE-MSCs Protect Neuronal Death in Parkinsons Disease Model.Int J Stem Cells. 2019 Mar 30;12(1):114-124. doi: 10.15283/ijsc18110. Int J Stem Cells. 2019. PMID: 30836725 Free PMC article.
References
-
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer’s disease. J Neuroimmunol. 1989;24:173–182. - PubMed
-
- Luber-Narod J, Rogers J. Immune system associated antigens expressed by cells of the human central nervous system. Neurosci Lett. 1988;94:17–22. - PubMed
-
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. - PubMed
-
- Eikelenboom P, Rosemuller AJ, Hoozemans JI, Veerhuis R, van Gool WA. Neuroinflammation and Alzheimer’s disease: Clinical and therapeutic implications. Alzheimer Dis Assoc Disord. 2000;14:S54–61. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
