

Human genomic disease variants: a neutral evolutionary explanation
- PMID: 22665443
- PMCID: PMC3409252
- DOI: 10.1101/gr.133702.111
Human genomic disease variants: a neutral evolutionary explanation
Abstract
Many perspectives on the role of evolution in human health include nonempirical assumptions concerning the adaptive evolutionary origins of human diseases. Evolutionary analyses of the increasing wealth of clinical and population genomic data have begun to challenge these presumptions. In order to systematically evaluate such claims, the time has come to build a common framework for an empirical and intellectual unification of evolution and modern medicine. We review the emerging evidence and provide a supporting conceptual framework that establishes the classical neutral theory of molecular evolution (NTME) as the basis for evaluating disease- associated genomic variations in health and medicine. For over a decade, the NTME has already explained the origins and distribution of variants implicated in diseases and has illuminated the power of evolutionary thinking in genomic medicine. We suggest that a majority of disease variants in modern populations will have neutral evolutionary origins (previously neutral), with a relatively smaller fraction exhibiting adaptive evolutionary origins (previously adaptive). This pattern is expected to hold true for common as well as rare disease variants. Ultimately, a neutral evolutionary perspective will provide medicine with an informative and actionable framework that enables objective clinical assessment beyond convenient tendencies to invoke past adaptive events in human history as a root cause of human disease.
Figures
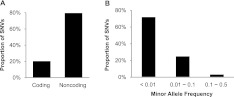
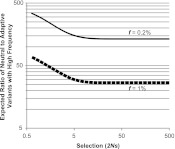
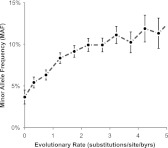
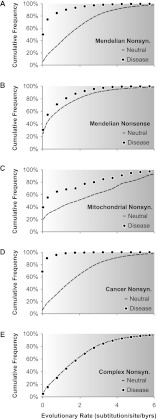
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous