

Molecular and Clinical Aspects of Angelman Syndrome
- PMID: 22670133
- PMCID: PMC3366701
- DOI: 10.1159/000328837
Molecular and Clinical Aspects of Angelman Syndrome
Abstract
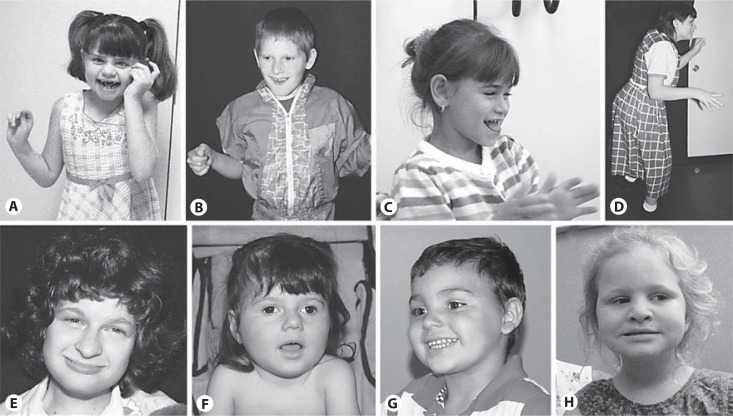
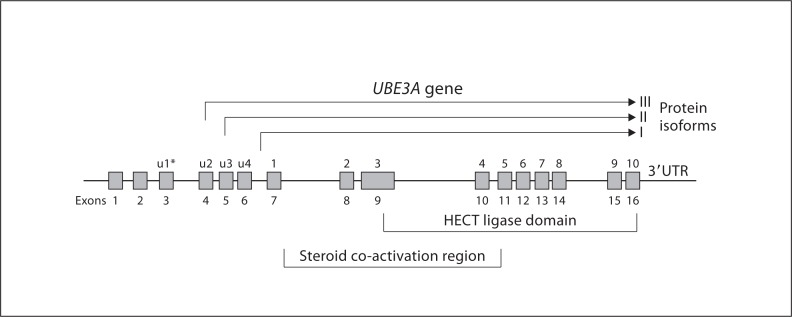
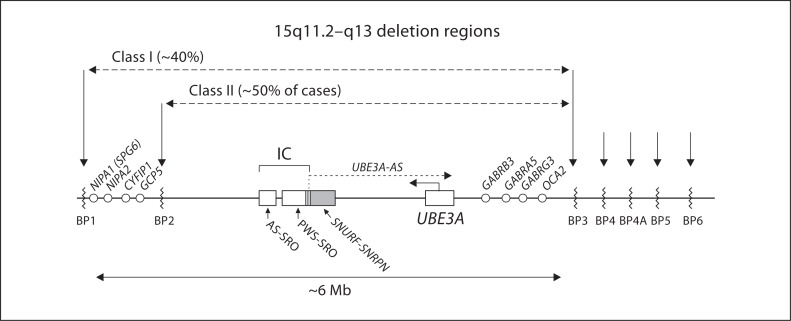
The Angelman syndrome is caused by disruption of the UBE3A gene and is clinically delineated by the combination of severe mental disability, seizures, absent speech, hypermotoric and ataxic movements, and certain remarkable behaviors. Those with the syndrome have a predisposition toward apparent happiness and paroxysms of laughter, and this finding helps distinguish Angelman syndrome from other conditions involving severe developmental handicap. Accurate diagnosis rests on a combination of clinical criteria and molecular and/or cytogenetic testing. Analysis of parent-specific DNA methylation imprints in the critical 15q11.2-q13 genomic region identifies 75-80% of all individuals with the syndrome, including those with cytogenetic deletions, imprinting center defects and paternal uniparental disomy. In the remaining group, UBE3A sequence analysis identifies an additional percentage of patients, but 5-10% will remain who appear to have the major clinical phenotypic features but do not have any identifiable genetic abnormalities. Genetic counseling for recurrence risk is complicated because multiple genetic mechanisms can disrupt the UBE3A gene, and there is also a unique inheritance pattern associated with UBE3A imprinting. Angelman syndrome is a prototypical developmental syndrome due to its remarkable behavioral phenotype and because UBE3A is so crucial to normal synaptic function and neural plasticity.
Figures
References
-
- Abaied L, Trabelsi M, Chaabouni M, Kharrat M, Kraoua L, et al. A novel UBE3A truncating mutation in large Tunisian Angelman syndrome pedigree. Am J Med Genet A. 2009;152A:141–146. - PubMed
-
- Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, et al. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet. 1997;17:75–78. - PubMed
-
- Angelman H. ‘Puppet' children. A report of three cases. Dev Med Child Neurol. 1965;7:681–688.
-
- Arn PH, Williams CA, Zori RT, Driscoll DJ, Rosenblatt DS. Methylenetetrahydrofolate reductase deficiency in a patient with phenotypic findings of Angelman syndrome. Am J Med Genet. 1998;77:198–200. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
