

CDKL5-Related Disorders: From Clinical Description to Molecular Genetics
- PMID: 22670135
- PMCID: PMC3366705
- DOI: 10.1159/000331333
CDKL5-Related Disorders: From Clinical Description to Molecular Genetics
Abstract
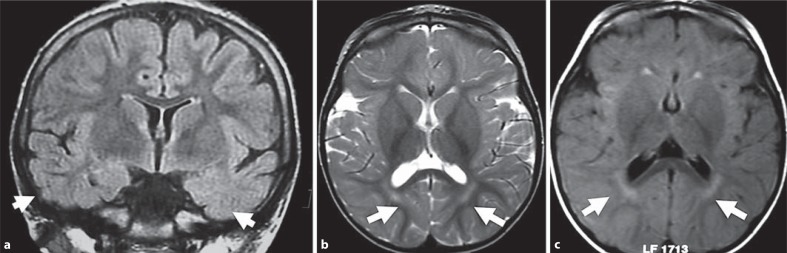
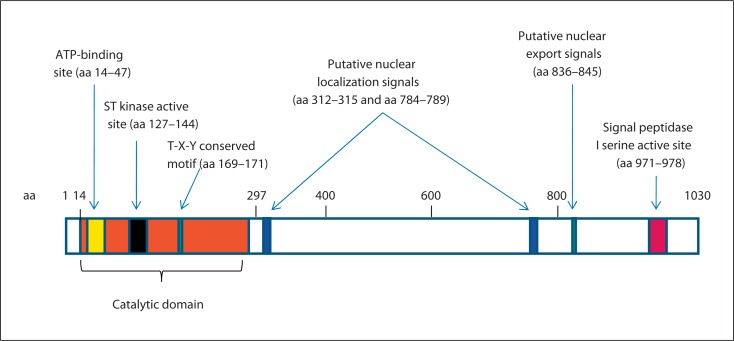
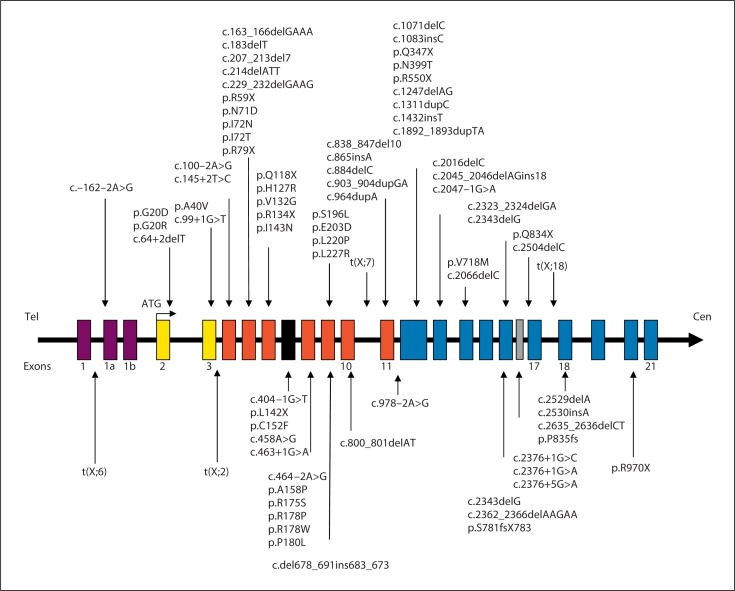
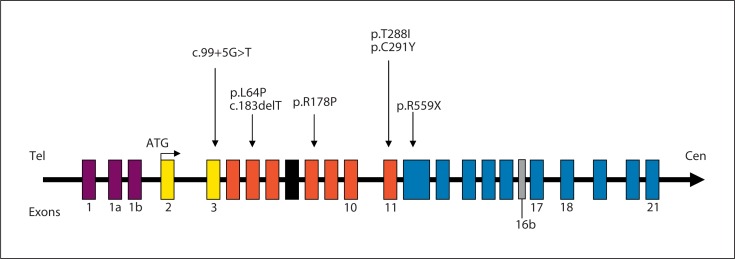
Mutations in the cyclin-dependent kinase-like 5 gene (CDKL5) have been described in girls with Rett-like features and early-onset epileptic encephalopathy including infantile spasms. To date, with more than 80 reported cases, the phenotype of CDKL5-related encephalopathy is better defined. The main features consist of early-onset seizures starting before 5 months of age, severe mental retardation with absent speech and Rett-like features such as hand stereotypies and deceleration of head growth. On the other hand, neuro-vegetative signs and developmental regression are rare in CDKL5 mutation patients. The CDKL5 gene encodes a serine threonine kinase protein which is characterized by a catalytic domain and a long C-terminal extension involved in the regulation of the catalytic activity of CDKL5 and in the sub-nuclear localization of the protein. To our knowledge, more than 70 different point mutations have been described including missense mutations within the catalytic domain, nonsense mutations causing the premature termination of the protein distributed in the entire open reading frame, splice variants, and frameshift mutations. Additionally, CDKL5 mutations have recently been described in 7 males with a more severe epileptic encephalopathy and a worse outcome compared to female patients. Finally, about 23 male and female patients have been identified with gross rearrangements encompassing all or part of the CDKL5 gene, with a phenotype reminiscent of CDKL5-related encephalopathy combined with dysmorphic features. Even if recent data clearly indicate that CDKL5 plays an important role in brain function, the protein remains largely uncharacterized. Phenotype-genotype correlation is additionally hampered by the relatively small number of patients described.
Figures
References
-
- Arts WF. CDKL5 gene-related epileptic encephalopathy: electroclinical findings in the first year of life. Dev Med Child Neurol. 2011;53:296–297. - PubMed
-
- Artuso R, Mencarelli MA, Polli R, Sartori S, Ariani F, et al. Early-onset seizure variant of Rett syndrome: definition of the clinical diagnostic criteria. Brain Dev. 2010;32:17–24. - PubMed
-
- Bahi-Buisson N, Nectoux J, Rosas-Vargas R, Milh M, Boddaert N, et al. Key clinical features to identify girls with CDKL5 mutations. Brain. 2008a;131:2647–2661. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
