

Adult Phenotypes in Angelman- and Rett-Like Syndromes
- PMID: 22670143
- PMCID: PMC3366698
- DOI: 10.1159/000335661
Adult Phenotypes in Angelman- and Rett-Like Syndromes
Abstract
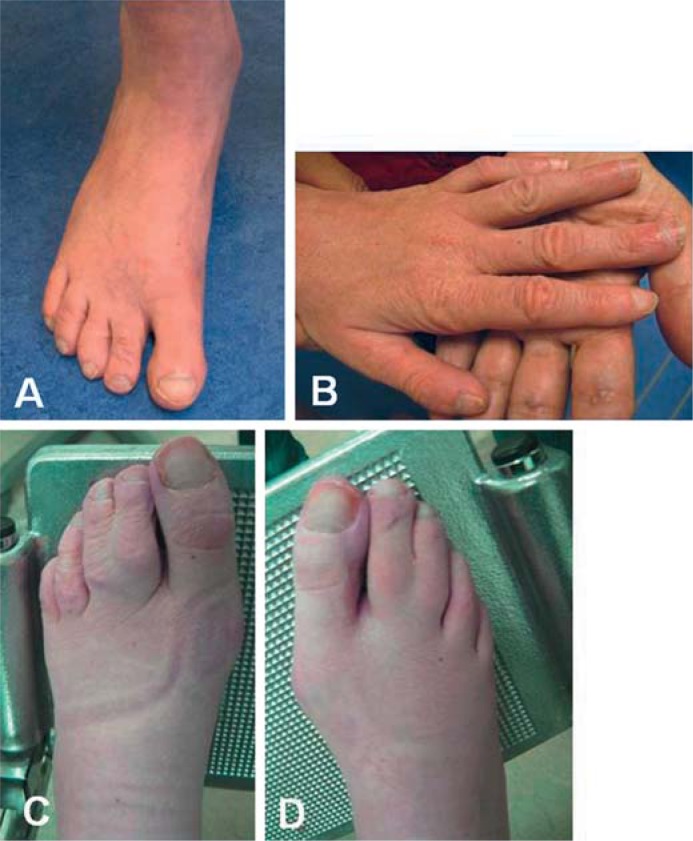
BACKGROUND: Angelman- and Rett-like syndromes share a range of clinical characteristics, including intellectual disability (ID) with or without regression, epilepsy, infantile encephalopathy, postnatal microcephaly, features of autism spectrum disorder, and variable other neurological symptoms. The phenotypic spectrum generally has been well studied in children; however, evolution of the phenotypic spectrum into adulthood has been documented less extensively. To obtain more insight into natural course and prognosis of these syndromes with respect to developmental, medical, and socio-behavioral outcomes, we studied the phenotypes of 9 adult patients who were recently diagnosed with 6 different Angelman- and Rett-like syndromes. METHODS: All these patients were ascertained during an ongoing cohort study involving a systematic clinical genetic diagnostic evaluation of over 250, mainly adult patients with ID of unknown etiology. RESULTS: We describe the evolution of the phenotype in adults with EHMT1, TCF4, MECP2, CDKL5, and SCN1A mutations and 22qter deletions and also provide an overview of previously published adult cases with similar diagnoses. CONCLUSION: These data are highly valuable in adequate management and follow-up of patients with Angelman- and Rett-like syndromes and accurate counseling of their family members. Furthermore, they will contribute to recognition of these syndromes in previously undiagnosed adult patients.
Figures

Similar articles
-
If not Angelman, what is it? A review of Angelman-like syndromes.Am J Med Genet A. 2014 Apr;164A(4):975-92. doi: 10.1002/ajmg.a.36416. Am J Med Genet A. 2014. PMID: 24779060 Review.
-
[Clinical usefulness of serial EEG examinations in the diagnostic of hereditary epileptic encephalopathies case of severe epileptic encephalopathy type 2].Przegl Lek. 2010;67(9):757-61. Przegl Lek. 2010. PMID: 21387820 Polish.
-
Rett syndrome: a study of the face.Am J Med Genet A. 2011 Jul;155A(7):1563-7. doi: 10.1002/ajmg.a.34027. Epub 2011 May 27. Am J Med Genet A. 2011. PMID: 21626673
-
Rett syndrome: significant clinical overlap with Angelman syndrome but not with methylation status.J Child Neurol. 1998 Sep;13(9):448-51. doi: 10.1177/088307389801300907. J Child Neurol. 1998. PMID: 9733292
-
Genetic disorders associated with postnatal microcephaly.Am J Med Genet C Semin Med Genet. 2014 Jun;166C(2):140-55. doi: 10.1002/ajmg.c.31400. Epub 2014 May 16. Am J Med Genet C Semin Med Genet. 2014. PMID: 24839169 Review.
Cited by
-
Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency.Mol Autism. 2013 Jun 11;4(1):18. doi: 10.1186/2040-2392-4-18. Mol Autism. 2013. PMID: 23758760 Free PMC article.
-
KCNQ2 encephalopathy manifesting with Rett-like features: A follow-up into adulthood.Neurol Genet. 2020 Sep 8;6(5):e510. doi: 10.1212/NXG.0000000000000510. eCollection 2020 Oct. Neurol Genet. 2020. PMID: 33134511 Free PMC article. No abstract available.
-
Clinical features and gene mutational spectrum of CDKL5-related diseases in a cohort of Chinese patients.BMC Med Genet. 2014 Feb 25;15:24. doi: 10.1186/1471-2350-15-24. BMC Med Genet. 2014. PMID: 24564546 Free PMC article.
-
Disordered breathing in a Pitt-Hopkins syndrome model involves Phox2b-expressing parafacial neurons and aberrant Nav1.8 expression.Nat Commun. 2021 Oct 13;12(1):5962. doi: 10.1038/s41467-021-26263-2. Nat Commun. 2021. PMID: 34645823 Free PMC article.
-
Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice.Proc Natl Acad Sci U S A. 2012 Dec 26;109(52):21516-21. doi: 10.1073/pnas.1216988110. Epub 2012 Dec 10. Proc Natl Acad Sci U S A. 2012. PMID: 23236174 Free PMC article.
References
-
- Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia. 2010;51:1043–1052. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Anderlid BM, Schoumans J, Annerén G, Tapia-Paez I, Dumanski J, et al. FISH-mapping of a 100-kb terminal 22q13 deletion. Hum Genet. 2002;110:439–443. - PubMed
LinkOut - more resources
Full Text Sources
