

Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells
- PMID: 22672192
- PMCID: PMC3441405
- DOI: 10.1186/1471-2164-13-220
Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells
Abstract
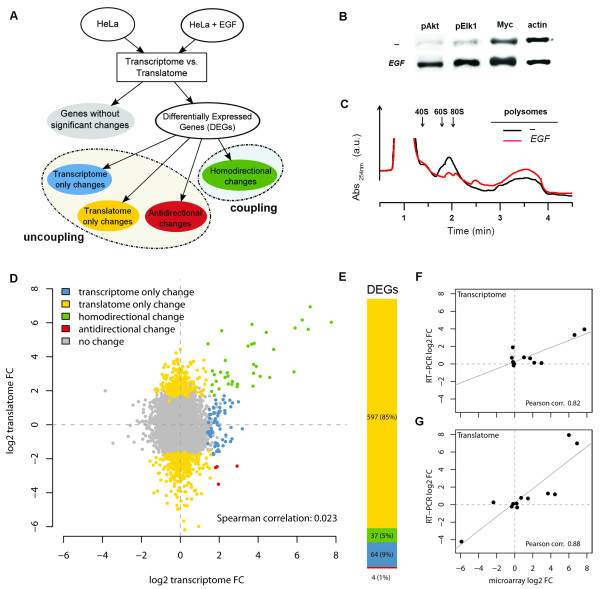
Background: The classical view on eukaryotic gene expression proposes the scheme of a forward flow for which fluctuations in mRNA levels upon a stimulus contribute to determine variations in mRNA availability for translation. Here we address this issue by simultaneously profiling with microarrays the total mRNAs (the transcriptome) and the polysome-associated mRNAs (the translatome) after EGF treatment of human cells, and extending the analysis to other 19 different transcriptome/translatome comparisons in mammalian cells following different stimuli or undergoing cell programs.
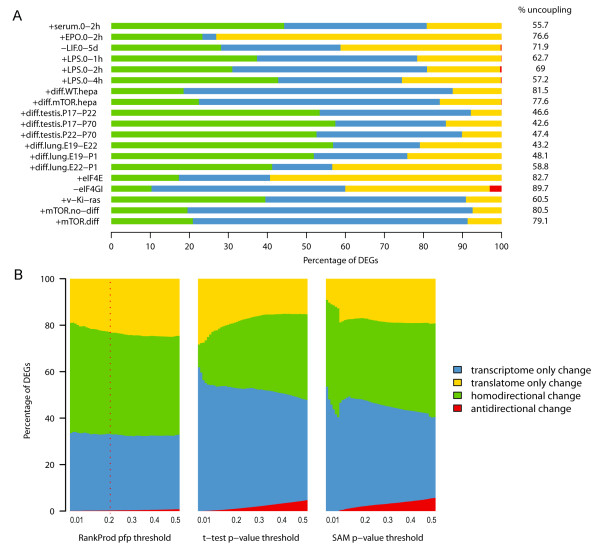
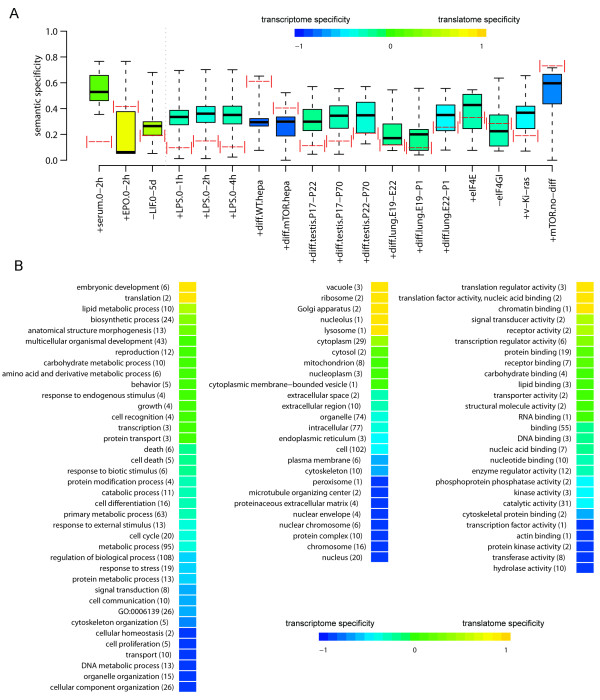
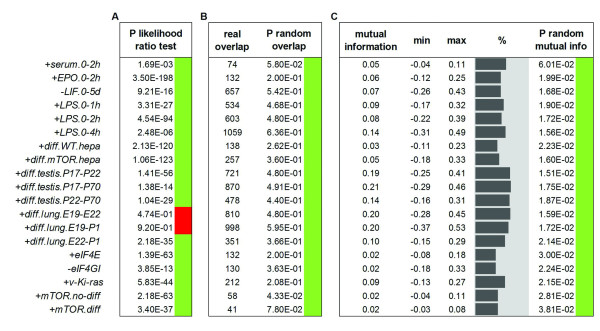
Results: Triggering of the EGF pathway results in an early induction of transcriptome and translatome changes, but 90% of the significant variation is limited to the translatome and the degree of concordant changes is less than 5%. The survey of other 19 different transcriptome/translatome comparisons shows that extensive uncoupling is a general rule, in terms of both RNA movements and inferred cell activities, with a strong tendency of translation-related genes to be controlled purely at the translational level. By different statistical approaches, we finally provide evidence of the lack of dependence between changes at the transcriptome and translatome levels.
Conclusions: We propose a model of diffused independency between variation in transcript abundances and variation in their engagement on polysomes, which implies the existence of specific mechanisms to couple these two ways of regulating gene expression.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
