

Sorafenib and pemetrexed toxicity in cancer cells is mediated via SRC-ERK signaling
- PMID: 22673740
- PMCID: PMC3679099
- DOI: 10.4161/cbt.20562
Sorafenib and pemetrexed toxicity in cancer cells is mediated via SRC-ERK signaling
Abstract
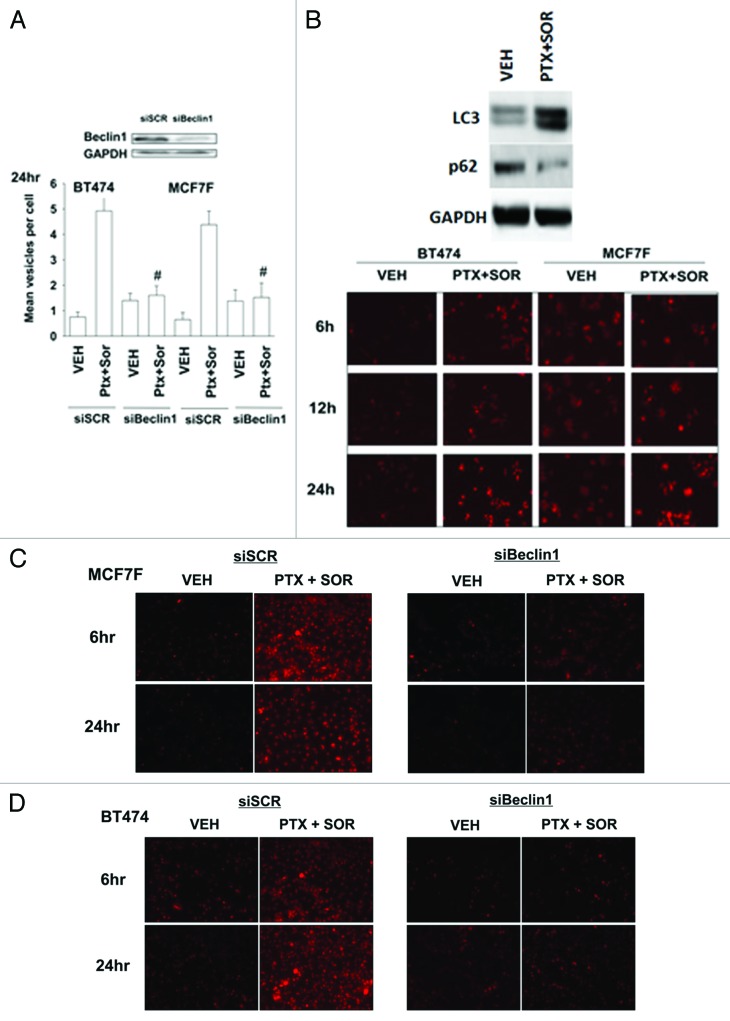
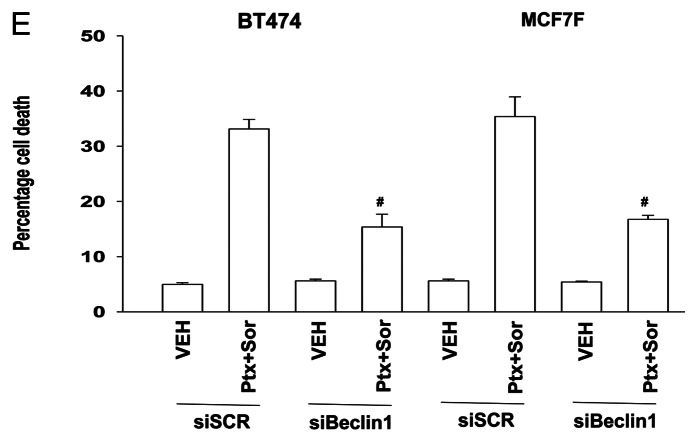
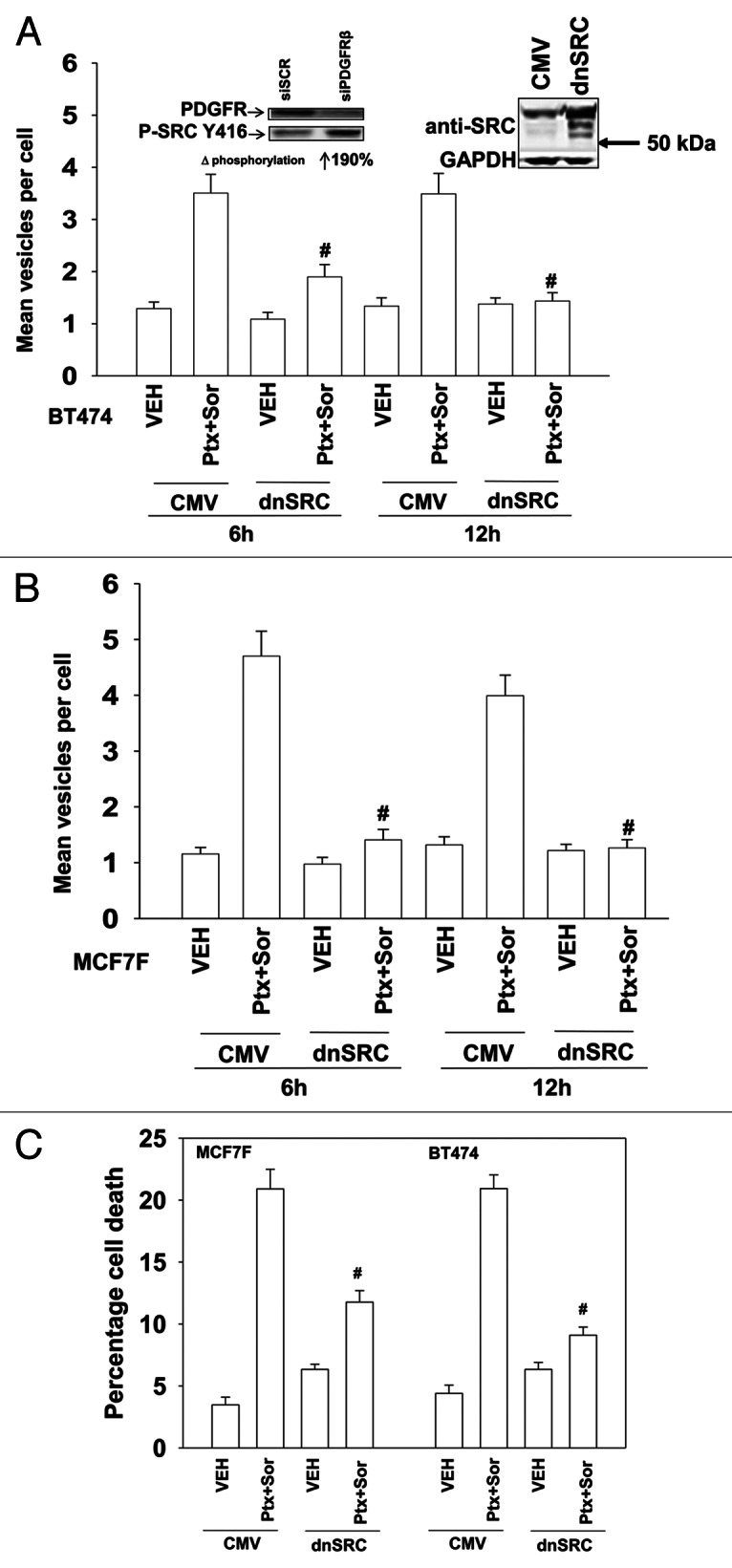
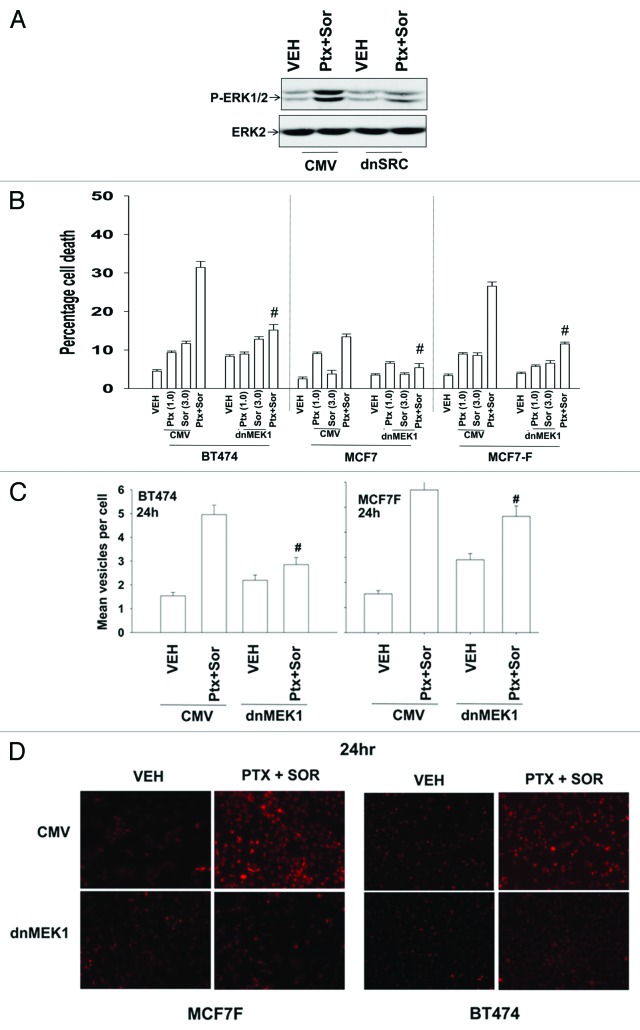
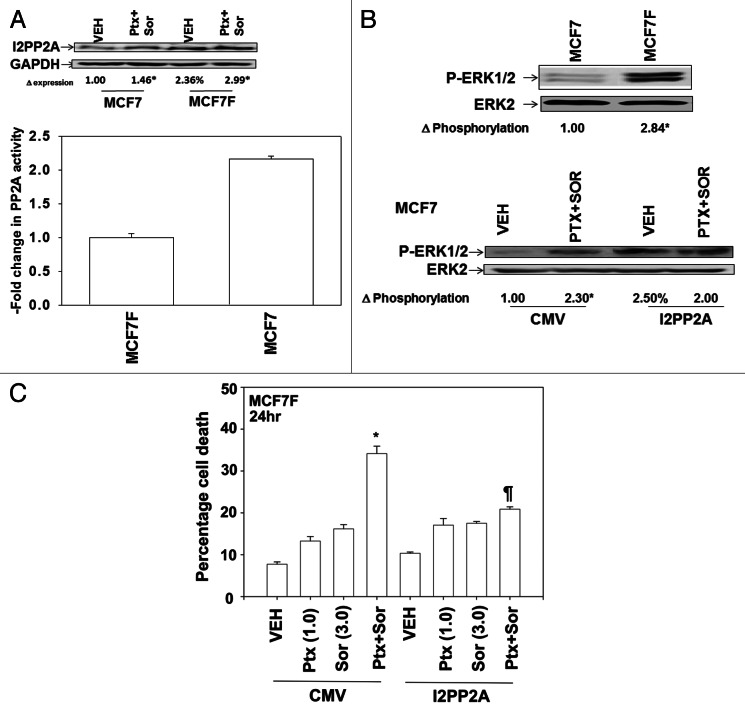
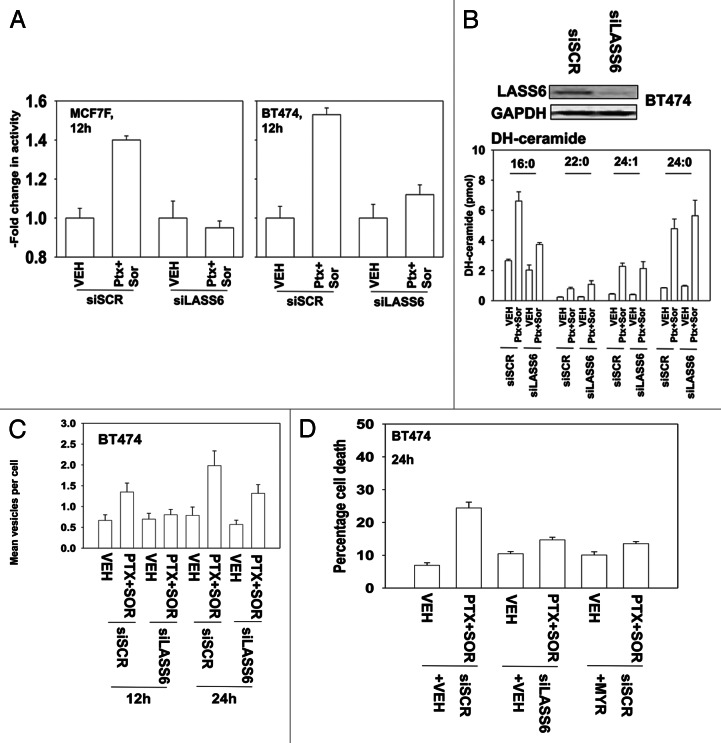
The present studies sought to further understand how the anti-folate pemetrexed and the multi-kinase inhibitor sorafenib interact to kill tumor cells. Sorafenib activated SRC, and via SRC the drug combination activated ERK1/2. Expression of dominant negative SRC or dominant negative MEK1 abolished drug-induced ERK1/2 activation, together with drug-induced autophagy, acidic lysosome formation, and tumor cell killing. Protein phosphatase 2A is an important regulator of the ERK1/2 pathway. Fulvestrant resistant MCF7 cells expressed higher levels of the PP2A inhibitor SET/I2PP2A, had lower endogenous PP2A activity, and had elevated basal ERK1/2 activity compared with their estrogen dependent counterparts. Overexpression of I2PP2A blocked drug-induced activation of ERK1/2 and tumor cell killing. PP2A can be directly activated by ceramide and SET/I2PP2A can be inhibited by ceramide. Inhibition of the de novo ceramide synthase pathway blocked drug-induced ceramide generation, PP2A activation and tumor cell killing. Collectively these findings demonstrate that ERK1/2 plays an essential role downstream of SRC in pemetrexed and sorafenib lethality and that PP2A plays an important role in regulating this process.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous