

A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance
- PMID: 22674929
- PMCID: PMC3378946
- DOI: 10.1136/bmjopen-2012-001124
A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance
Abstract
Objectives: To investigate the prospects of newly available benchtop sequencers to provide rapid whole-genome data in routine clinical practice. Next-generation sequencing has the potential to resolve uncertainties surrounding the route and timing of person-to-person transmission of healthcare-associated infection, which has been a major impediment to optimal management.
Design: The authors used Illumina MiSeq benchtop sequencing to undertake case studies investigating potential outbreaks of methicillin-resistant Staphylococcus aureus (MRSA) and Clostridium difficile.
Setting: Isolates were obtained from potential outbreaks associated with three UK hospitals.
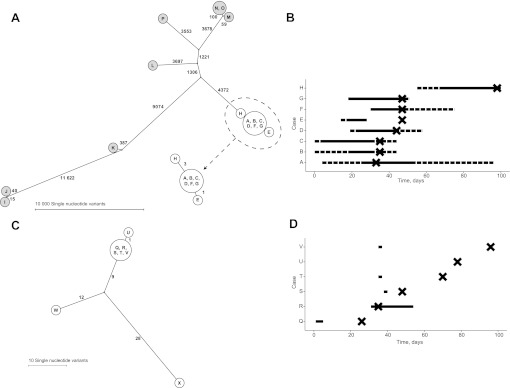
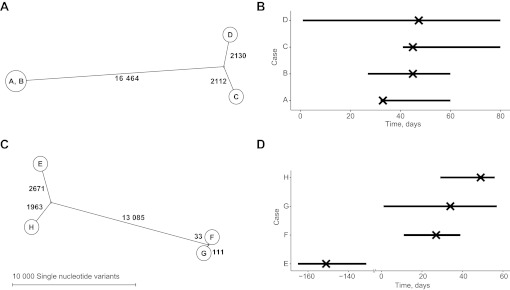
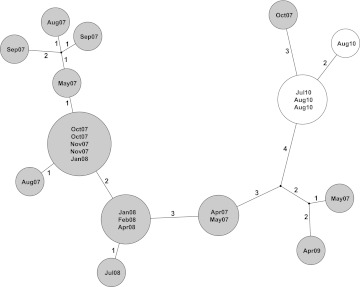
Participants: Isolates were sequenced from a cluster of eight MRSA carriers and an associated bacteraemia case in an intensive care unit, another MRSA cluster of six cases and two clusters of C difficile. Additionally, all C difficile isolates from cases over 6 weeks in a single hospital were rapidly sequenced and compared with local strain sequences obtained in the preceding 3 years.
Main outcome measure: Whole-genome genetic relatedness of the isolates within each epidemiological cluster.
Results: Twenty-six MRSA and 15 C difficile isolates were successfully sequenced and analysed within 5 days of culture. Both MRSA clusters were identified as outbreaks, with most sequences in each cluster indistinguishable and all within three single nucleotide variants (SNVs). Epidemiologically unrelated isolates of the same spa-type were genetically distinct (≥21 SNVs). In both C difficile clusters, closely epidemiologically linked cases (in one case sharing the same strain type) were shown to be genetically distinct (≥144 SNVs). A reconstruction applying rapid sequencing in C difficile surveillance provided early outbreak detection and identified previously undetected probable community transmission.
Conclusions: This benchtop sequencing technology is widely generalisable to human bacterial pathogens. The findings provide several good examples of how rapid and precise sequencing could transform identification of transmission of healthcare-associated infection and therefore improve hospital infection control and patient outcomes in routine clinical practice.
Conflict of interest statement
Figures

References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources