

Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling
- PMID: 22674974
- PMCID: PMC3421739
- DOI: 10.1128/JVI.00533-12
Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling
Abstract
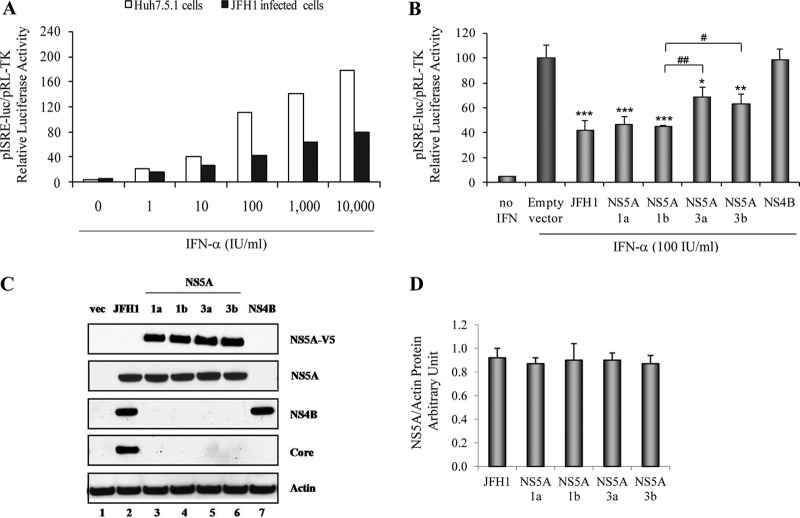
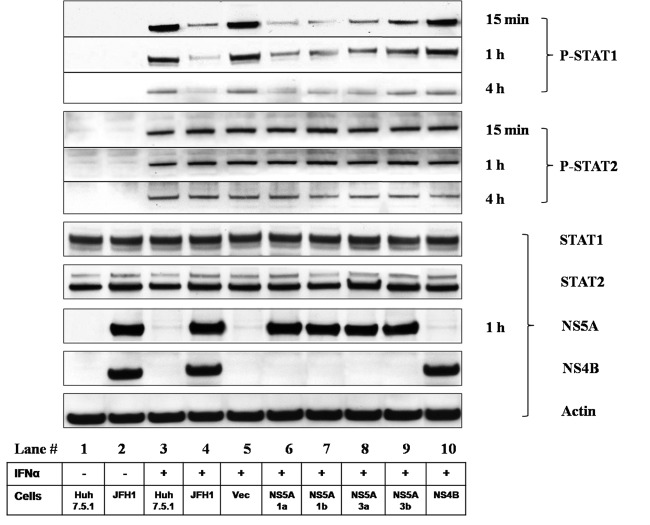
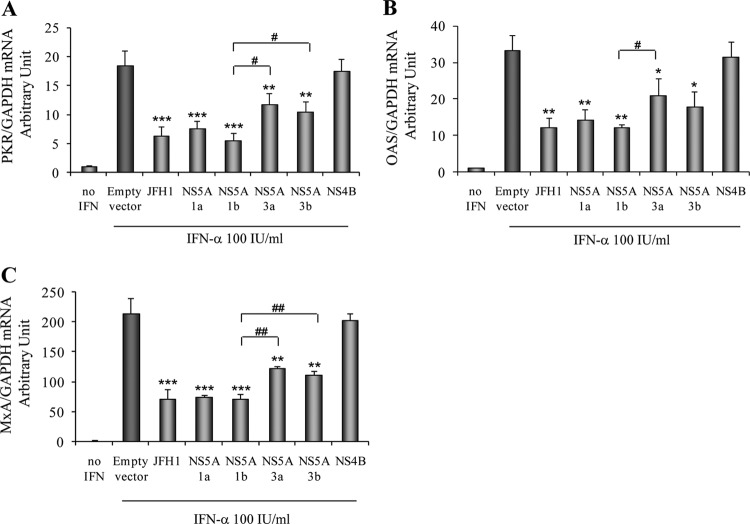
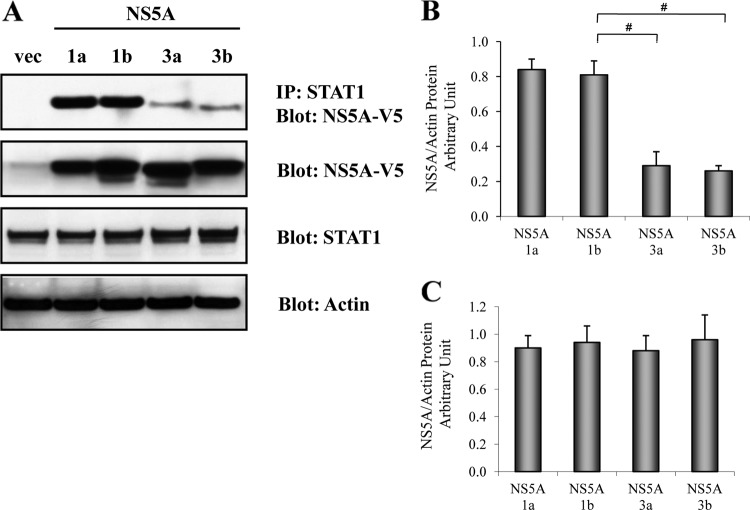
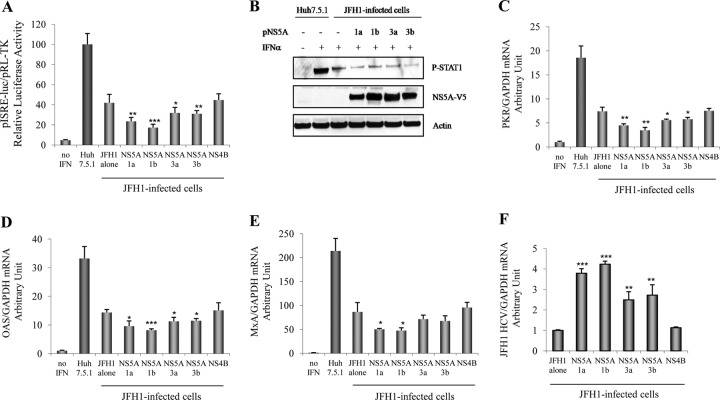
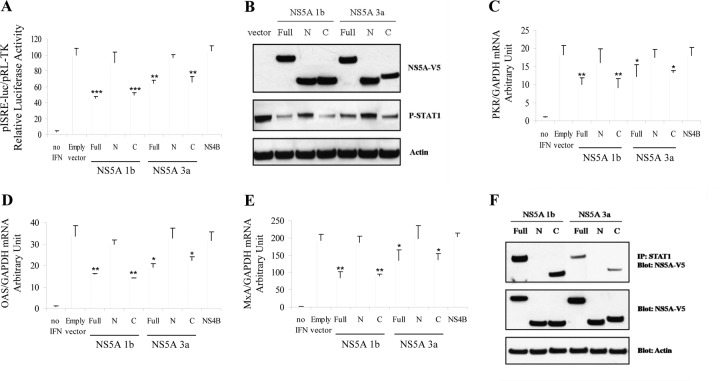
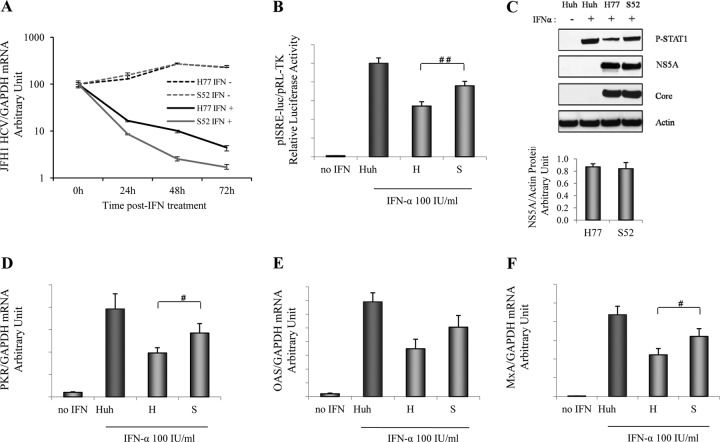
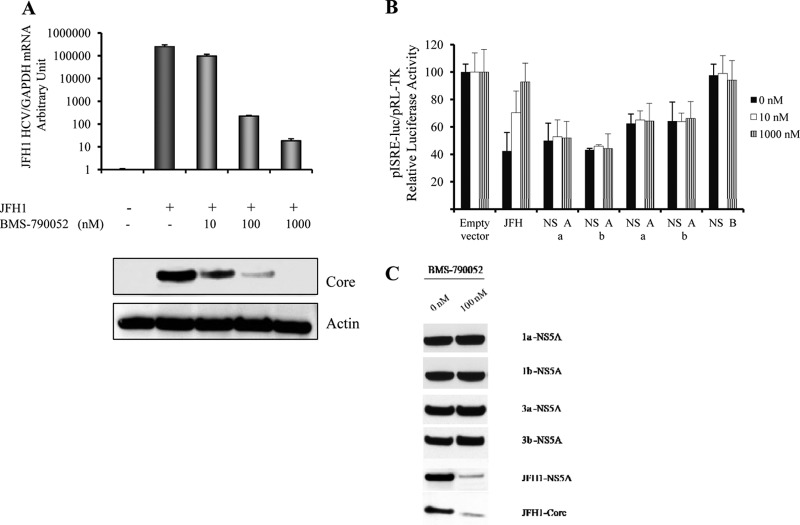
Responses to alpha interferon (IFN-α)-based treatment are dependent on both host and viral factors and vary markedly among patients infected with different hepatitis C virus (HCV) genotypes (GTs). Patients infected with GT3 viruses consistently respond better to IFN treatment than do patients infected with GT1 viruses. The mechanisms underlying this difference are not well understood. In this study, we sought to determine the effects of HCV NS5A proteins from different genotypes on IFN signaling. We found that the overexpression of either GT1 or GT3 NS5A proteins significantly inhibited IFN-induced IFN-stimulated response element (ISRE) signaling, phosphorylated STAT1 (P-STAT1) levels, and IFN-stimulated gene (ISG) expression compared to controls. GT1 NS5A protein expression exhibited stronger inhibitory effects on IFN signaling than did GT3 NS5A protein expression. Furthermore, GT1 NS5A bound to STAT1 with a higher affinity than did GT3 NS5A. Domain mapping revealed that the C-terminal region of NS5A conferred these inhibitory effects on IFN signaling. The overexpression of HCV NS5A increased HCV replication levels in JFH1-infected cells through the further reduction of levels of P-STAT1, ISRE signaling, and downstream ISG responses. We demonstrated that the overexpression of GT1 NS5A proteins resulted in less IFN responsiveness than did the expression of GT3 NS5A proteins through stronger binding to STAT1. We confirmed that GT1 NS5A proteins exerted stronger IFN signaling inhibition than did GT3 NS5A proteins in an infectious recombinant JFH1 virus. The potent antiviral NS5A inhibitor BMS-790052 did not block NS5A-mediated IFN signaling suppression in an overexpression model, suggesting that NS5A's contributions to replication are independent of its subversive action on IFN. We propose a model in which the binding of the C-terminal region of NS5A to STAT1 leads to decreased levels of P-STAT1, ISRE signaling, and ISG transcription and, ultimately, to preferential GT1 resistance to IFN treatment.
Figures
Similar articles
-
HCV NS5A inhibits interferon-alpha signaling through suppression of STAT1 phosphorylation in hepatocyte-derived cell lines.J Hepatol. 2007 May;46(5):759-67. doi: 10.1016/j.jhep.2006.11.013. Epub 2006 Dec 14. J Hepatol. 2007. PMID: 17275127
-
Hepatitis C virus non-structural 5A abrogates signal transducer and activator of transcription-1 nuclear translocation induced by IFN-alpha through dephosphorylation.World J Gastroenterol. 2007 Aug 14;13(30):4080-4. doi: 10.3748/wjg.v13.i30.4080. World J Gastroenterol. 2007. PMID: 17696225 Free PMC article.
-
[Effect of HCV NS5A on STAT1 phosphorylation and nuclear translocation induced by IFN alpha-2b].Zhonghua Gan Zang Bing Za Zhi. 2006 Dec;14(12):894-7. Zhonghua Gan Zang Bing Za Zhi. 2006. PMID: 17196131 Chinese.
-
Understanding the molecular mechanism(s) of hepatitis C virus (HCV) induced interferon resistance.Infect Genet Evol. 2013 Oct;19:113-9. doi: 10.1016/j.meegid.2013.06.025. Epub 2013 Jul 5. Infect Genet Evol. 2013. PMID: 23831932 Review.
-
The nonstructural NS5A protein of hepatitis C virus: an expanding, multifunctional role in enhancing hepatitis C virus pathogenesis.J Biomed Sci. 2002 May-Jun;9(3):187-97. doi: 10.1007/BF02256065. J Biomed Sci. 2002. PMID: 12065893 Review.
Cited by
-
Acetaldehyde accelerates HCV-induced impairment of innate immunity by suppressing methylation reactions in liver cells.Am J Physiol Gastrointest Liver Physiol. 2015 Oct 1;309(7):G566-77. doi: 10.1152/ajpgi.00183.2015. Epub 2015 Aug 6. Am J Physiol Gastrointest Liver Physiol. 2015. PMID: 26251470 Free PMC article.
-
What role (if any) does the highly conserved CSB-PGBD3 fusion protein play in Cockayne syndrome?Mech Ageing Dev. 2013 May-Jun;134(5-6):225-33. doi: 10.1016/j.mad.2013.01.001. Epub 2013 Jan 28. Mech Ageing Dev. 2013. PMID: 23369858 Free PMC article. Review.
-
Fludarabine as an Adjuvant Improves Newcastle Disease Virus-Mediated Antitumor Immunity in Hepatocellular Carcinoma.Mol Ther Oncolytics. 2019 Mar 27;13:22-34. doi: 10.1016/j.omto.2019.03.004. eCollection 2019 Jun 28. Mol Ther Oncolytics. 2019. PMID: 31011625 Free PMC article.
-
Hepatitis C, innate immunity and alcohol: friends or foes?Biomolecules. 2015 Feb 5;5(1):76-94. doi: 10.3390/biom5010076. Biomolecules. 2015. PMID: 25664450 Free PMC article. Review.
-
Atypical Porcine Pestivirus Circulation and Molecular Evolution within an Affected Swine Herd.Viruses. 2020 Sep 25;12(10):1080. doi: 10.3390/v12101080. Viruses. 2020. PMID: 32992946 Free PMC article.
References
-
- Barth H, Liang TJ, Baumert TF. 2006. Hepatitis C virus entry: molecular biology and clinical implications. Hepatology 44:527–535 - PubMed
-
- Chen L, et al. 2005. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology 128:1437–1444 - PubMed
-
- Darnell JE, Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421 - PubMed
-
- Ferenci P, et al. 2005. Predicting sustained virological responses in chronic hepatitis C patients treated with peginterferon alfa-2a (40 KD)/ribavirin. J. Hepatol. 43:425–433 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
