

KOMODO: a web tool for detecting and visualizing biased distribution of groups of homologous genes in monophyletic taxa
- PMID: 22675073
- PMCID: PMC3394310
- DOI: 10.1093/nar/gks490
KOMODO: a web tool for detecting and visualizing biased distribution of groups of homologous genes in monophyletic taxa
Abstract
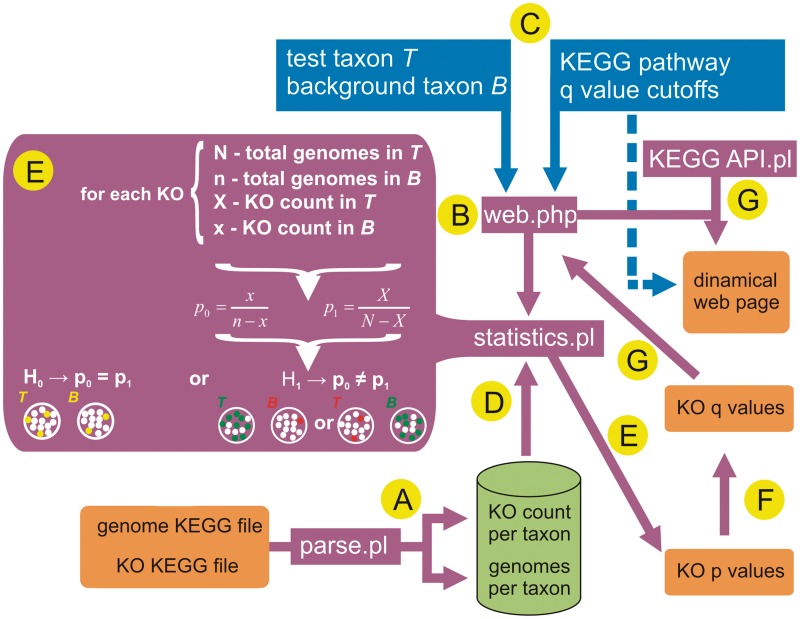
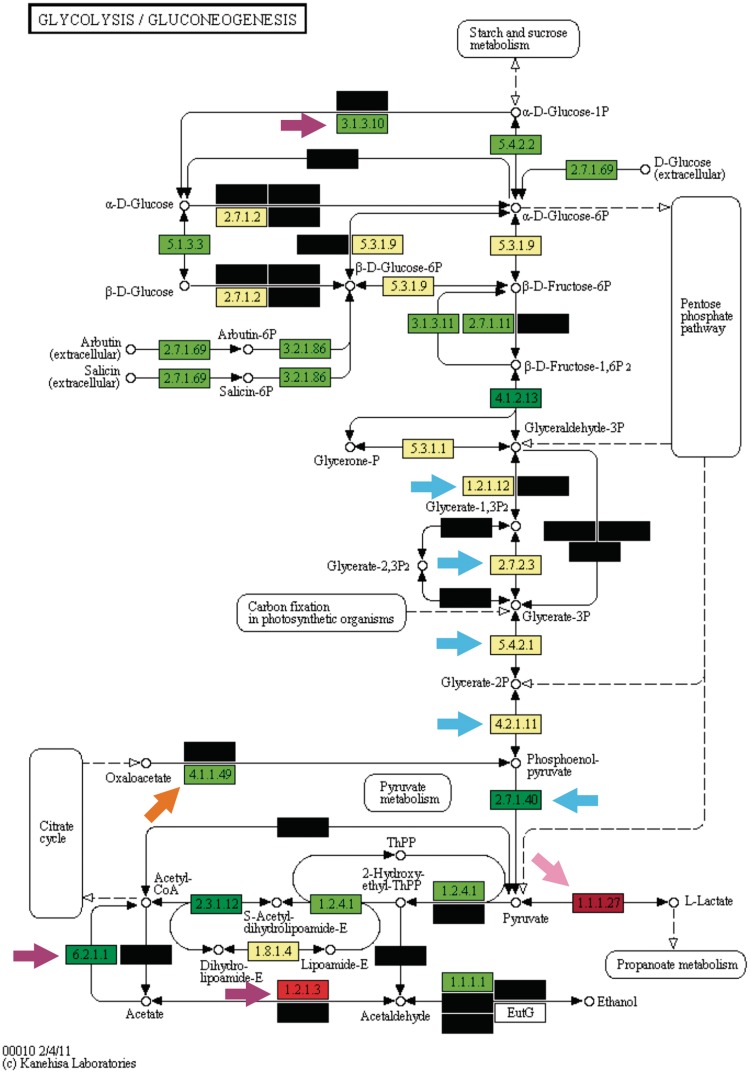
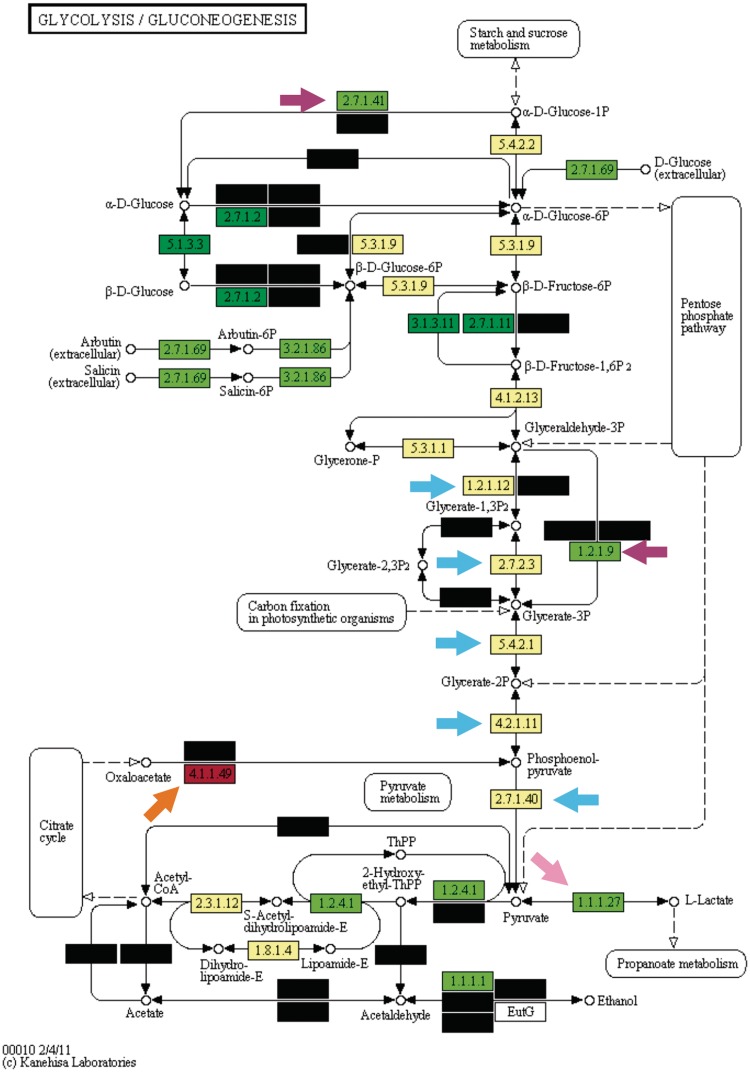
The enrichment analysis is a standard procedure to interpret 'omics' experiments that generate large gene lists as outputs, such as transcriptomics and protemics. However, despite the huge success of enrichment analysis in these classes of experiments, there is a surprising lack of application of this methodology to survey other categories of large-scale biological data available. Here, we report Kegg Orthology enrichMent-Online DetectiOn (KOMODO), a web tool to systematically investigate groups of monophyletic genomes in order to detect significantly enriched groups of homologous genes in one taxon when compared with another. The results are displayed in their proper biochemical roles in a visual, explorative way, allowing users to easily formulate and investigate biological hypotheses regarding the taxonomical distribution of genomic elements. We validated KOMODO by analyzing portions of central carbon metabolism in two taxa extensively studied regarding their carbon metabolism profile (Enterobacteriaceae family and Lactobacillales order). Most enzymatic activities significantly biased were related to known key metabolic traits in these taxa, such as the distinct fates of pyruvate (the known tendency of lactate production in Lactobacillales and its complete oxidation in Enterobacteriaceae), demonstrating that KOMODO could detect biologically meaningful differences in the frequencies of shared genomic elements among taxa. KOMODO is freely available at http://komodotool.org.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
