

Improved strategy for phylogenetic analysis of classical swine fever virus based on full-length E2 encoding sequences
- PMID: 22676246
- PMCID: PMC3403906
- DOI: 10.1186/1297-9716-43-50
Improved strategy for phylogenetic analysis of classical swine fever virus based on full-length E2 encoding sequences
Abstract
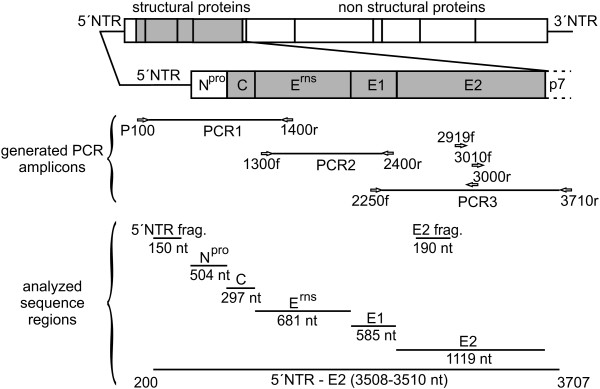
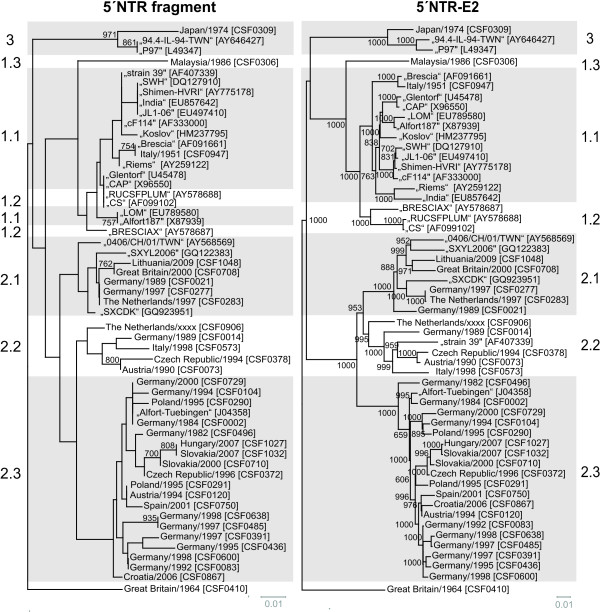
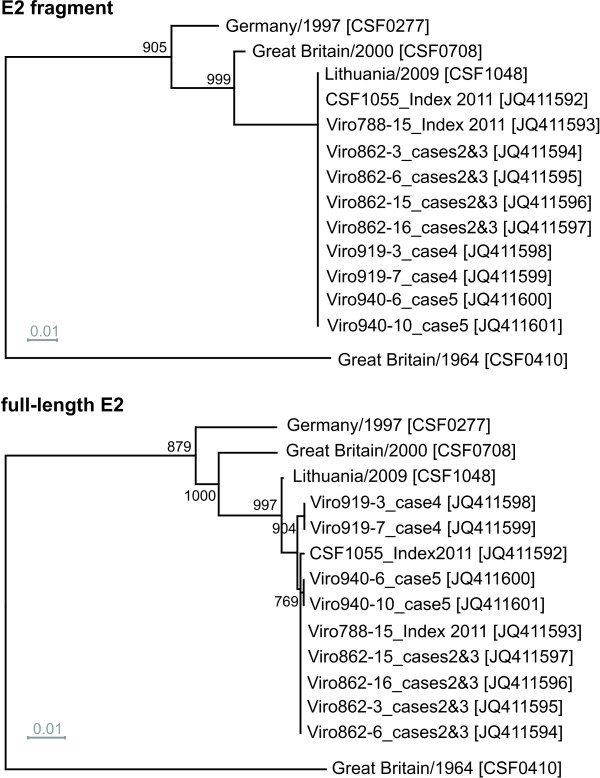
Molecular epidemiology has proven to be an essential tool in the control of classical swine fever (CSF) and its use has significantly increased during the past two decades. Phylogenetic analysis is a prerequisite for virus tracing and thus allows implementing more effective control measures. So far, fragments of the 5´NTR (150 nucleotides, nt) and the E2 gene (190 nt) have frequently been used for phylogenetic analyses. The short sequence lengths represent a limiting factor for differentiation of closely related isolates and also for confidence levels of proposed CSFV groups and subgroups. In this study, we used a set of 33 CSFV isolates in order to determine the nucleotide sequences of a 3508-3510 nt region within the 5´ terminal third of the viral genome. Including 22 additional sequences from GenBank database different regions of the genome, comprising the formerly used short 5´NTR and E2 fragments as well as the genomic regions encoding the individual viral proteins Npro, C, Erns, E1, and E2, were compared with respect to variability and suitability for phylogenetic analysis. Full-length E2 encoding sequences (1119 nt) proved to be most suitable for reliable and statistically significant phylogeny and analyses revealed results as good as obtained with the much longer entire 5´NTR-E2 sequences. This strategy is therefore recommended by the EU and OIE Reference Laboratory for CSF as it provides a solid and improved basis for CSFV molecular epidemiology. Finally, the power of this method is illustrated by the phylogenetic analysis of closely related CSFV isolates from a recent outbreak in Lithuania.
Figures
References
-
- Depner KR, Strebelow G, Staubach C, Kramer M, Teuffert J, Botcher L, Hoffmann B, Beer M, Greiser-Wilke I, Mettenleiter T. Case report: the significance of genotyping for the epidemiological tracing of classical swine fever (CSF) Dtsch Tierarztl Wochenschr. 2006;113:159–162. - PubMed
-
- Vilcek S, Herring AJ, Herring JA, Nettleton PF, Lowings JP, Paton DJ. Pestiviruses isolated from pigs, cattle and sheep can be allocated into at least three genogroups using polymerase chain reaction and restriction endonuclease analysis. Arch Virol. 1994;136:309–323. doi: 10.1007/BF01321060. - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
