

Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C
- PMID: 22676771
- PMCID: PMC3465012
- DOI: 10.1186/1750-1172-7-36
Miglustat therapy in the French cohort of paediatric patients with Niemann-Pick disease type C
Abstract
Background: Niemann-Pick disease type C (NP-C) is a rare neurovisceral lysosomal lipid storage disease characterized by progressive neurological deterioration. Published data on the use of miglustat in paediatric patients in clinical practice settings are limited. We report findings from a prospective open-label study in the French paediatric NP-C cohort.
Methods: Data on all paediatric NP-C patients treated with miglustat in France between October 2006 and December 2010 were compiled. All patients had a confirmed diagnosis of NP-C, and received miglustat therapy according to manufacturer's recommendations. Pre-treatment and follow-up assessments were conducted according to a standardized protocol.
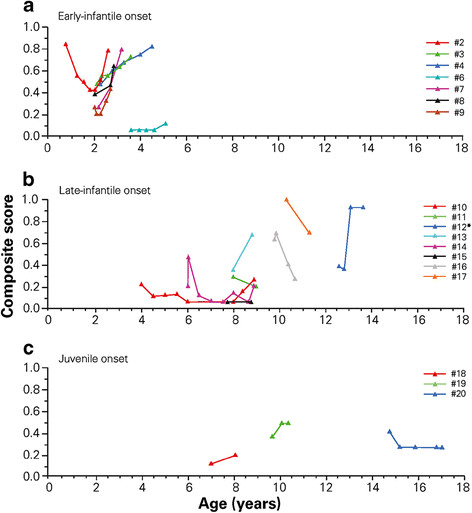
Results: Twenty children were enrolled; 19 had NPC1 gene mutations and 1 had NPC2 gene mutations. The median age at diagnosis was 1.5 years, and the median age at miglustat initiation was 6.0 years. Eight NPC1 patients had the early-infantile, eight had the late-infantile, and three had the juvenile-onset forms of NP-C. A history of hepatosplenomegaly and/or other cholestatic symptoms was recorded in all 8 early-infantile onset patients, 3/8 late-infantile patients, and 1/3 juvenile onset patients. Brain imaging indicated white matter abnormalities in most patients. The median (range) duration of miglustat therapy was 1.3 (0.6-2.3) years in early-infantile, 1.0 (0.8-5.0) year in late-infantile, and 1.0 (0.6-2.5) year in juvenile onset patients. NP-C disability scale scores indicated either stabilization or improvement of neurological manifestations in 1/8, 6/8, and 1/3 NPC1 patients in these subgroups, respectively. There were no correlations between brain imaging findings and disease course. Mild-to-moderate gastrointestinal disturbances were frequent during the first 3 months of miglustat therapy, but were easily managed with dietary modifications and/or anti-propulsive medication.
Conclusions: Miglustat can improve or stabilize neurological manifestations in paediatric patients with the late-infantile and juvenile-onset forms of NP-C. Among early-infantile onset patients, a shorter delay between neurological disease onset and miglustat initiation was associated with an initial better therapeutic outcome in one patient, but miglustat did not seem to modify overall disease course in this subgroup. More experience is required with long-term miglustat therapy in early-infantile onset patients treated from the very beginning of neurological manifestations.
Figures

References
-
- Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, Pineda M, Sedel F, Topcu M, Vanier MT. et al.Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009;98:152–165. - PubMed
-
- Wraith JE, Guffon N, Rohrbach M, Hwu WL, Korenke GC, Bembi B, Luzy C, Giorgino R, Sedel F. Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol Genet Metab. 2009;98:250–254. - PubMed
-
- Pineda M, Wraith JE, Mengel E, Sedel F, Hwu WL, Rohrbach M, Bembi B, Walterfang M, Korenke GC, Marquardt T. et al.Miglustat in patients with Niemann-Pick disease Type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab. 2009;98:243–249. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
