

Two-stage extreme phenotype sequencing design for discovering and testing common and rare genetic variants: efficiency and power
- PMID: 22678112
- PMCID: PMC3558993
- DOI: 10.1159/000337300
Two-stage extreme phenotype sequencing design for discovering and testing common and rare genetic variants: efficiency and power
Abstract
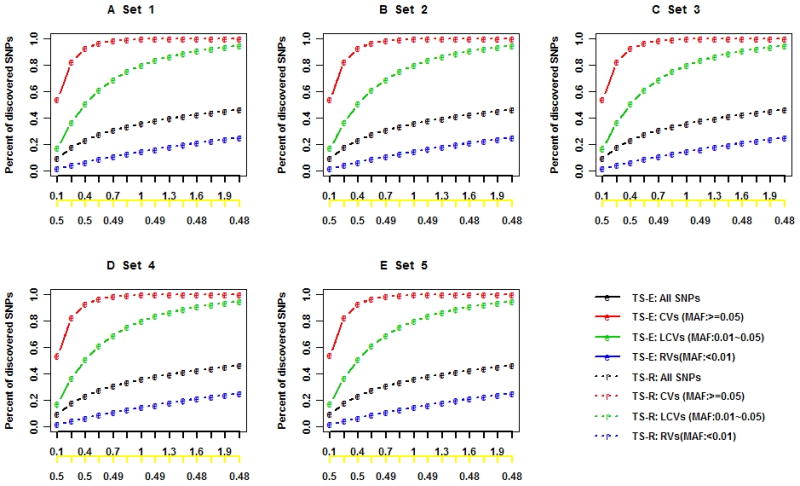
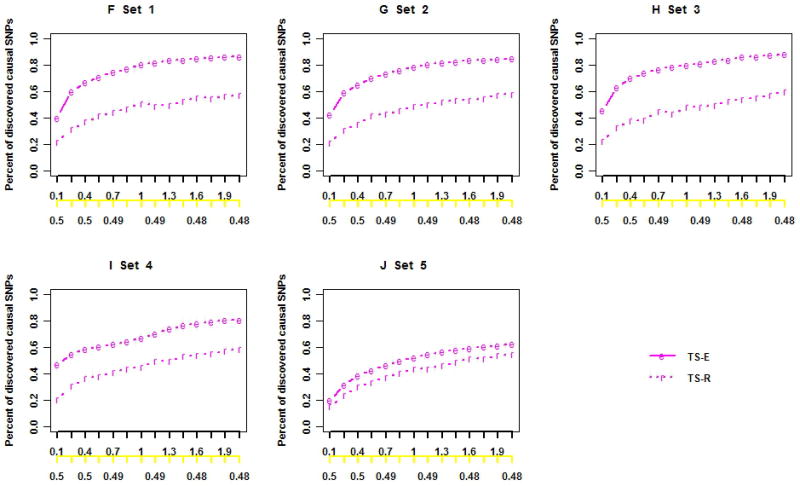
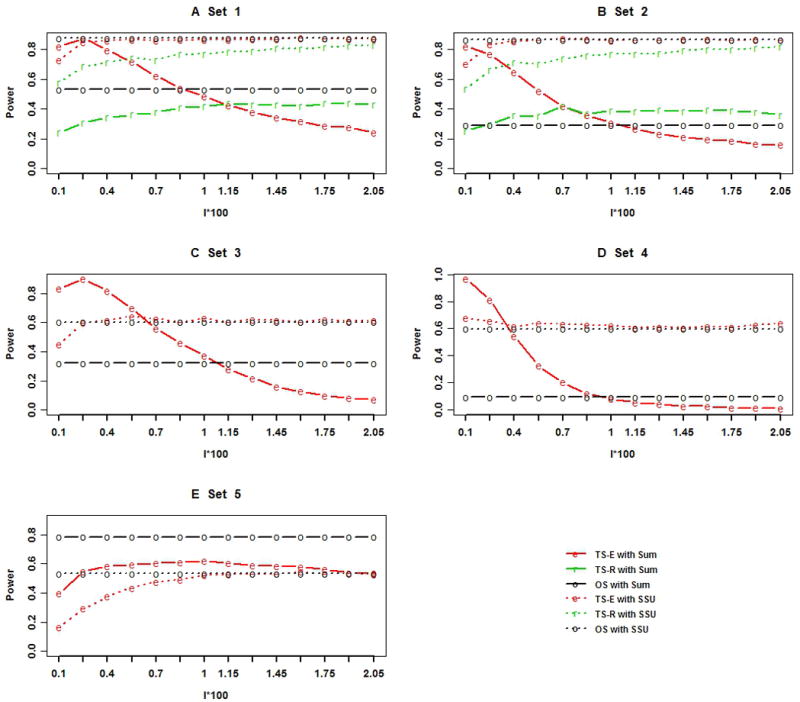
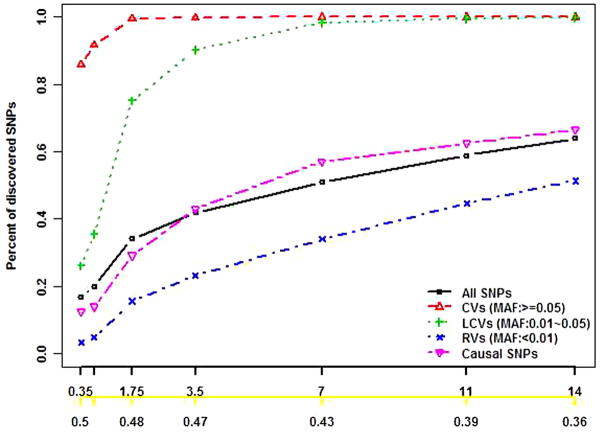
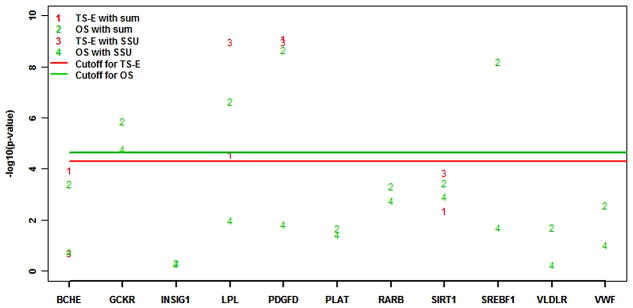
Next-generation sequencing technology provides an unprecedented opportunity to identify rare susceptibility variants. It is not yet financially feasible to perform whole-genome sequencing on a large number of subjects, and a two-stage design has been advocated to be a practical option. In stage I, variants are discovered by sequencing the whole genomes of a small number of carefully selected individuals. In stage II, the discovered variants of a large number of individuals are genotyped to assess associations. Individuals with extreme phenotypes are typically selected in stage I. Using simulated data for unrelated individuals, we explore two important aspects of this two-stage design: the efficiency of discovering common and rare single-nucleotide polymorphisms (SNPs) in stage I and the impact of incomplete SNP discovery in stage I on the power of testing associations in stage II. We applied a sum test and a sum of squared score test for gene-based association analyses evaluating the power of the two-stage design. We obtained the following results from extensive simulation studies and analysis of the GAW17 dataset. When individuals with trait values more extreme than the 99.7-99th quantile were included in stage I, the two-stage design could achieve the same power as or even higher than the one-stage design if the rare causal variants had large effect sizes. In such design, fewer than half of the total SNPs including more than half of the causal SNPs were discovered, which included nearly all SNPs with minor allele frequencies (MAFs) ≥5%, more than half of the SNPs with MAFs between 1% and 5%, and fewer than half of the SNPs with MAFs <1%. Although a one-stage design may be preferable to identify multiple rare variants having small to moderate effect sizes, our observations support using the two-stage design as a cost-effective option for next-generation sequencing studies.
Copyright © 2012 S. Karger AG, Basel.
Figures
Similar articles
-
Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies.Genet Epidemiol. 2011 Dec;35(8):790-9. doi: 10.1002/gepi.20628. Epub 2011 Sep 15. Genet Epidemiol. 2011. PMID: 21922541 Free PMC article.
-
A unified approach for allele frequency estimation, SNP detection and association studies based on pooled sequencing data using EM algorithms.BMC Genomics. 2013;14 Suppl 1(Suppl 1):S1. doi: 10.1186/1471-2164-14-S1-S1. Epub 2013 Jan 21. BMC Genomics. 2013. PMID: 23369070 Free PMC article.
-
Detection of common single nucleotide polymorphisms synthesizing quantitative trait association of rarer causal variants.Genome Res. 2011 Jul;21(7):1122-30. doi: 10.1101/gr.115832.110. Epub 2011 Mar 25. Genome Res. 2011. PMID: 21441355 Free PMC article.
-
Identifying rare variants associated with complex traits via sequencing.Curr Protoc Hum Genet. 2013 Jul;Chapter 1:Unit 1.26. doi: 10.1002/0471142905.hg0126s78. Curr Protoc Hum Genet. 2013. PMID: 23853079 Free PMC article. Review.
-
Linkage analysis in the next-generation sequencing era.Hum Hered. 2011;72(4):228-36. doi: 10.1159/000334381. Epub 2011 Dec 23. Hum Hered. 2011. PMID: 22189465 Free PMC article. Review.
Cited by
-
Cancer pharmacogenomics: strategies and challenges.Nat Rev Genet. 2013 Jan;14(1):23-34. doi: 10.1038/nrg3352. Epub 2012 Nov 27. Nat Rev Genet. 2013. PMID: 23183705 Free PMC article. Review.
-
Phenotypic extremes in rare variant study designs.Eur J Hum Genet. 2016 Jun;24(6):924-30. doi: 10.1038/ejhg.2015.197. Epub 2015 Sep 9. Eur J Hum Genet. 2016. PMID: 26350511 Free PMC article.
-
A Systematic Review of Extreme Phenotype Strategies to Search for Rare Variants in Genetic Studies of Complex Disorders.Genes (Basel). 2020 Aug 25;11(9):987. doi: 10.3390/genes11090987. Genes (Basel). 2020. PMID: 32854191 Free PMC article.
-
Low-, high-coverage, and two-stage DNA sequencing in the design of the genetic association study.Genet Epidemiol. 2017 Apr;41(3):187-197. doi: 10.1002/gepi.22015. Epub 2016 Nov 4. Genet Epidemiol. 2017. PMID: 27813156 Free PMC article.
-
Comparison of mixed model based approaches for correcting for population substructure with application to extreme phenotype sampling.BMC Genomics. 2022 Feb 4;23(1):98. doi: 10.1186/s12864-022-08297-y. BMC Genomics. 2022. PMID: 35120458 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
