

Homeostasis and the importance for a balance between AKT/mTOR activity and intracellular signaling
- PMID: 22680924
- PMCID: PMC3414727
- DOI: 10.2174/092986712801661130
Homeostasis and the importance for a balance between AKT/mTOR activity and intracellular signaling
Abstract
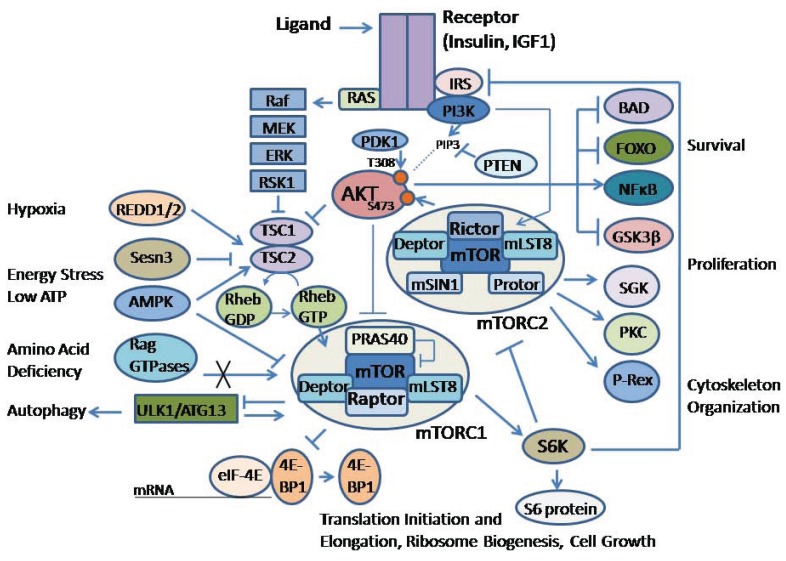
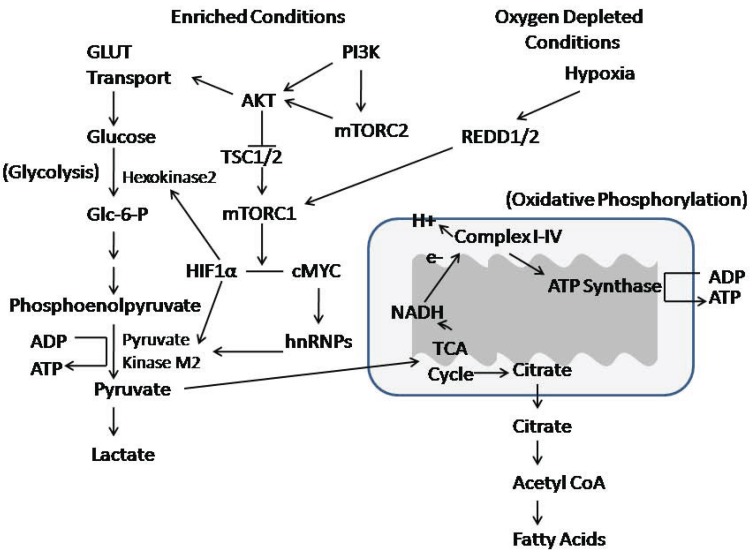
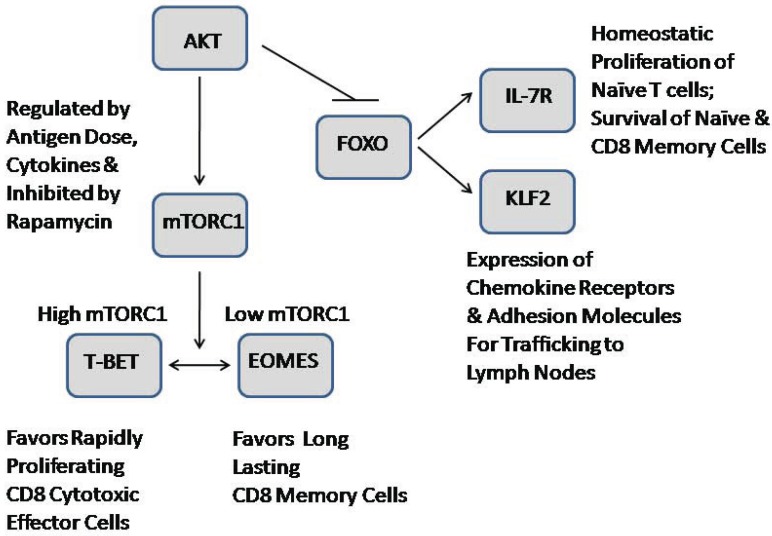
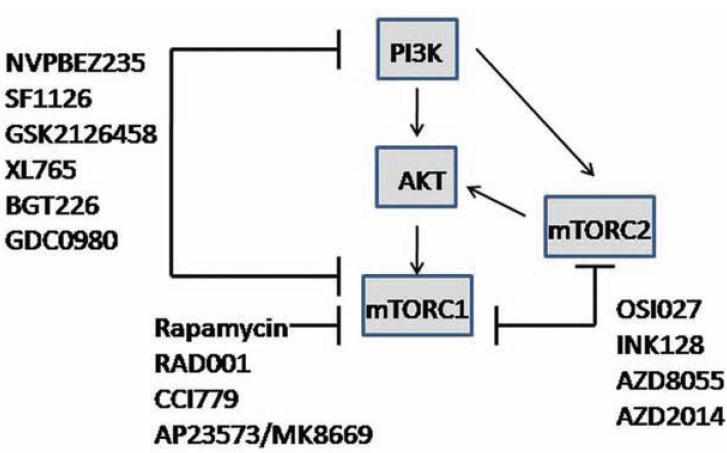
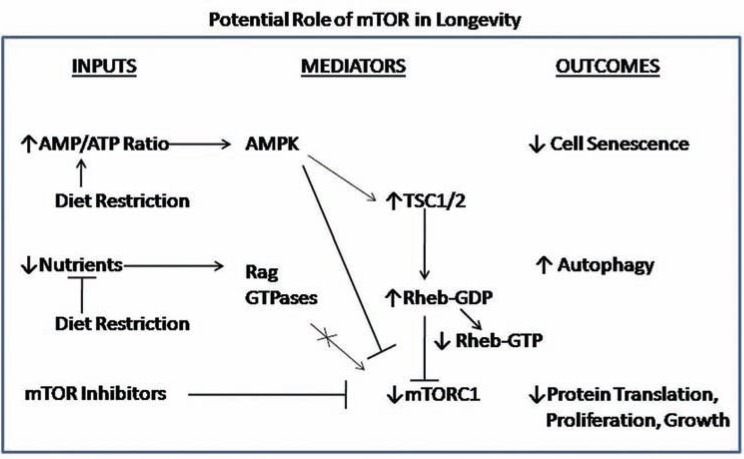
The AKT family of serine threonine kinases is of critical importance with regard to growth factor signaling, cell proliferation, survival and oncogenesis. Engagement of signaling receptors induces the lipid kinase, phosphatidylinositol 3-kinase (PI3K), which enables the activation of AKT. Responsive to the PI3K/AKT pathway is the mammalian target of rapamycin (mTOR), a major effector that is specifically implicated in the regulation of cell growth as a result of nutrient availability and cellular bioenergetics. These kinases mediate the activity of a multitude of intracellular signaling molecules and intersect with multiple pathways that regulate cellular processes. Elucidating the role of AKT/mTOR in metabolism and in hallmark signaling pathways that are aberrantly affected in cancer has provided a solid foundation of discoveries. From this, new research directions are emerging with regard to the role of AKT/mTOR in diabetes and T cell-mediated immunity. As a result, a new perspective is developing in how AKT/mTOR functions within intracellular signaling pathways to maintain cellular homeostasis. An appreciation is emerging that altered equilibrium of AKT/mTOR pathways contributes to disease and malignancy. Such new insights may lead to novel intervention strategies that may be useful to reprogram or reset the balance of intracellular signaling.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous