

Improving the accuracy of macromolecular structure refinement at 7 Å resolution
- PMID: 22681901
- PMCID: PMC3380535
- DOI: 10.1016/j.str.2012.04.020
Improving the accuracy of macromolecular structure refinement at 7 Å resolution
Abstract
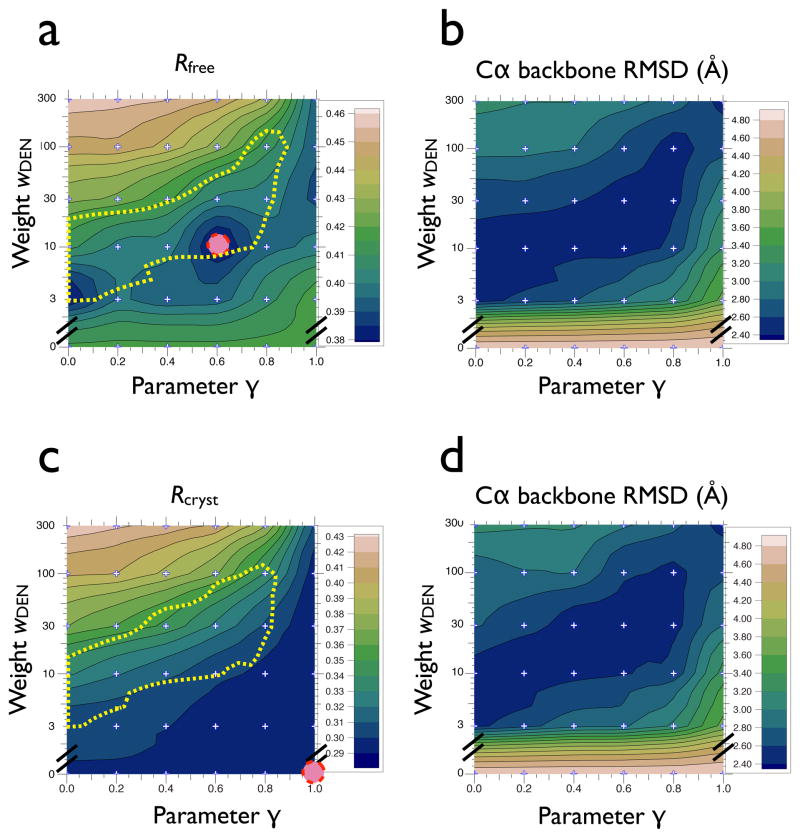
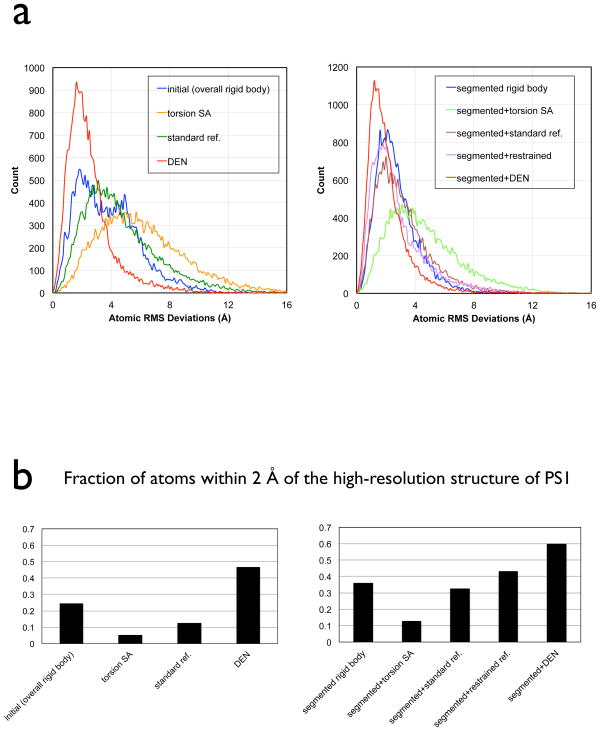
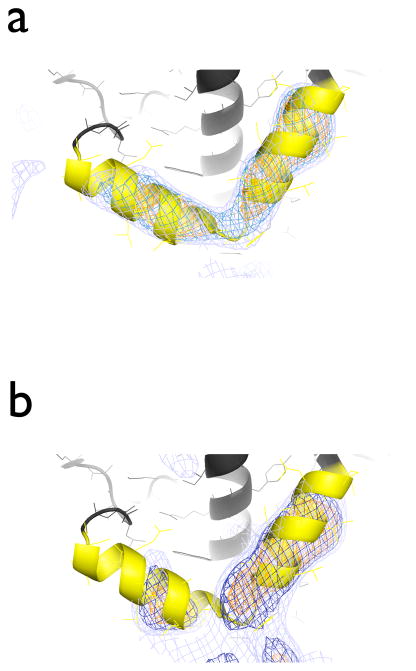
In X-ray crystallography, molecular replacement and subsequent refinement is challenging at low resolution. We compared refinement methods using synchrotron diffraction data of photosystem I at 7.4 Å resolution, starting from different initial models with increasing deviations from the known high-resolution structure. Standard refinement spoiled the initial models, moving them further away from the true structure and leading to high R(free)-values. In contrast, DEN refinement improved even the most distant starting model as judged by R(free), atomic root-mean-square differences to the true structure, significance of features not included in the initial model, and connectivity of electron density. The best protocol was DEN refinement with initial segmented rigid-body refinement. For the most distant initial model, the fraction of atoms within 2 Å of the true structure improved from 24% to 60%. We also found a significant correlation between R(free) values and the accuracy of the model, suggesting that R(free) is useful even at low resolution.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures

References
-
- Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. - PubMed
-
- Brunger AT. Free R value: cross-validation in crystallography. Methods Enzymol. 1997;277:366–396. - PubMed
-
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
