

Comparative epigenomic annotation of regulatory DNA
- PMID: 22682255
- PMCID: PMC3372872
- DOI: 10.1016/j.cell.2012.04.029
Comparative epigenomic annotation of regulatory DNA
Abstract
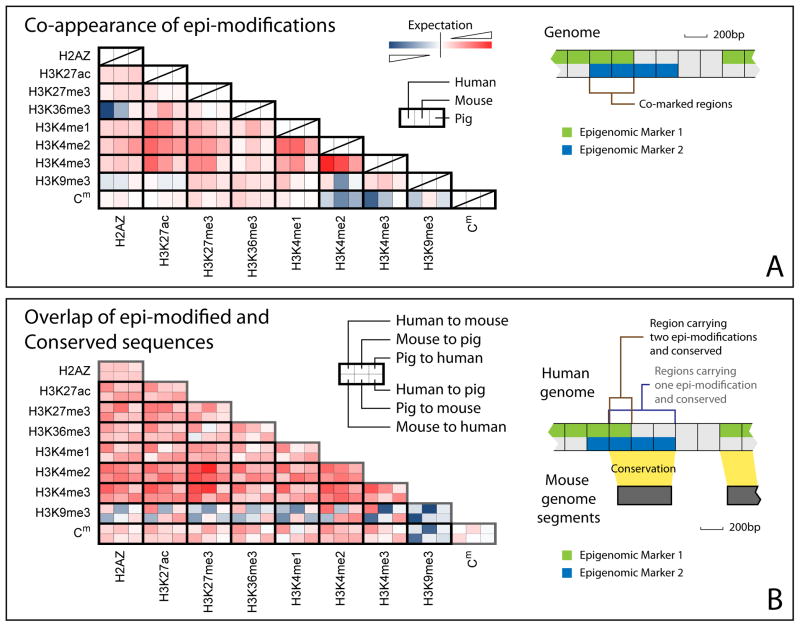
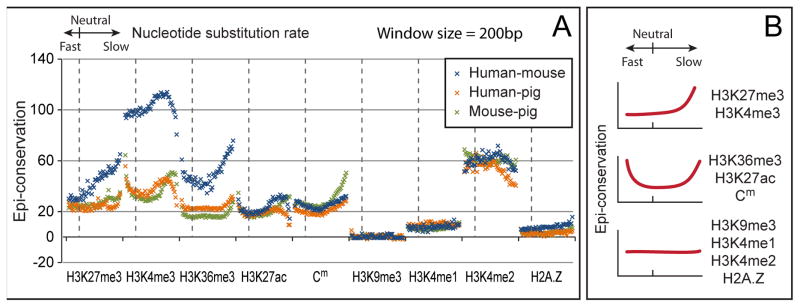
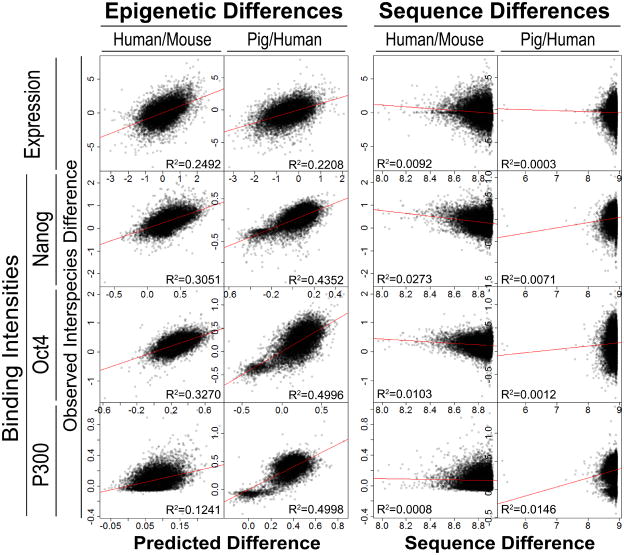
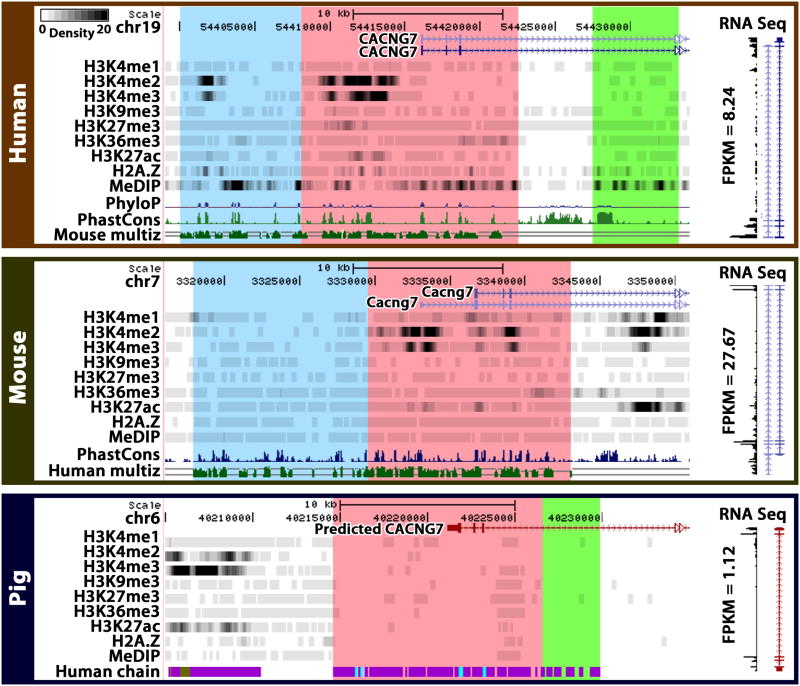
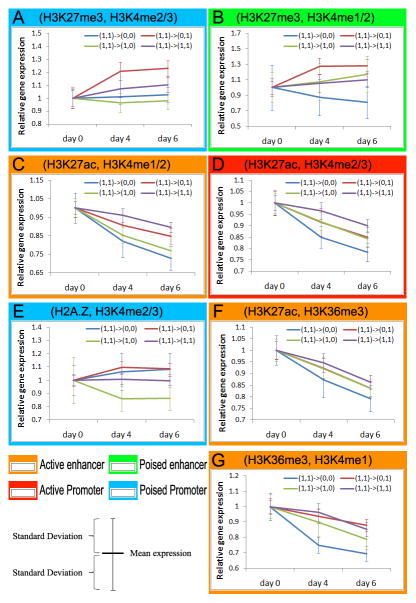
Despite the explosive growth of genomic data, functional annotation of regulatory sequences remains difficult. Here, we introduce "comparative epigenomics"-interspecies comparison of DNA and histone modifications-as an approach for annotation of the regulatory genome. We measured in human, mouse, and pig pluripotent stem cells the genomic distributions of cytosine methylation, H2A.Z, H3K4me1/2/3, H3K9me3, H3K27me3, H3K27ac, H3K36me3, transcribed RNAs, and P300, TAF1, OCT4, and NANOG binding. We observed that epigenomic conservation was strong in both rapidly evolving and slowly evolving DNA sequences, but not in neutrally evolving sequences. In contrast, evolutionary changes of the epigenome and the transcriptome exhibited a linear correlation. We suggest that the conserved colocalization of different epigenomic marks can be used to discover regulatory sequences. Indeed, seven pairs of epigenomic marks identified exhibited regulatory functions during differentiation of embryonic stem cells into mesendoderm cells. Thus, comparative epigenomics reveals regulatory features of the genome that cannot be discerned from sequence comparisons alone.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures

References
-
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell. 2006;125:315–326. - PubMed
-
- Campos EI, Reinberg D. Histones: Annotating Chromatin. Annual Review of Genetics. 2009;43:559–599. - PubMed
-
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
