

Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis
- PMID: 22682420
- PMCID: PMC3446494
- DOI: 10.1186/ar3843
Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening disease of severe hyperinflammation caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting high amounts of inflammatory cytokines. It is a frequent manifestation in patients with predisposing genetic defects, but can occur secondary to various infectious, malignant, and autoimmune triggers in patients without a known genetic predisposition. Clinical hallmarks are prolonged fever, cytopenias, hepatosplenomegaly, and neurological symptoms, but atypical variants presenting with signs of chronic immunodeficiency are increasingly recognized. Impaired secretion of perforin is a key feature in several genetic forms of the disease, but not required for disease pathogenesis. Despite progress in diagnostics and therapy, mortality of patients with severe HLH is still above 40%. Reference treatment is an etoposide-based protocol, but new approaches are currently explored. Key for a favorable prognosis is the rapid identification of an underlying genetic cause, which has been facilitated by recent immunological and genetic advances. In patients with predisposing genetic disease, hematopoietic stem cell transplantation is performed increasingly with reduced intensity conditioning regimes. Current research aims at a better understanding of disease pathogenesis and evaluation of more targeted approaches to therapy, including anti-cytokine antibodies and gene therapy.
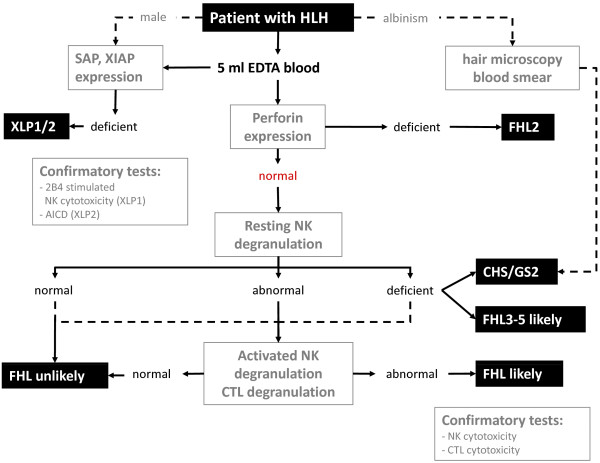
Figures



References
-
- Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, Ohta H, Fujisaki H, Kobayashi M, Sakata N, Kawa-Ha K, Okada S, Tawa A. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89:4100–4103. - PubMed
-
- Pachlopnik Schmid J, Cote M, Menager MM, Burgess A, Nehme N, Menasche G, Fischer A, de Saint Basile G. Inherited defects in lymphocyte cytotoxic activity. Immunol Rev. pp. 10–23. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
