

Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation
- PMID: 22683086
- PMCID: PMC3397269
- DOI: 10.1016/j.ajhg.2012.05.004
Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation
Abstract
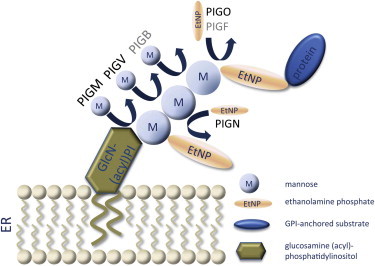
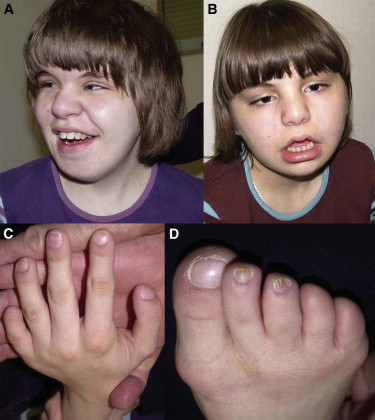
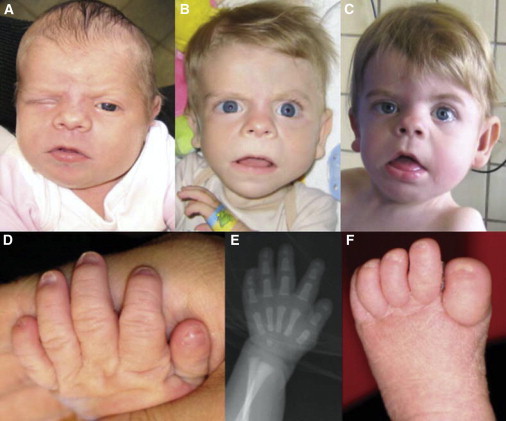
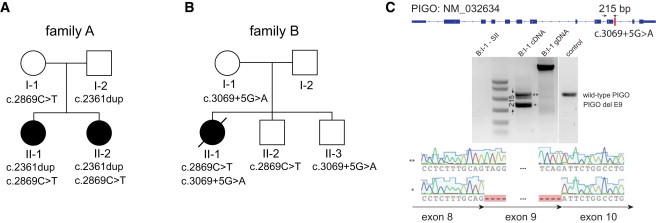
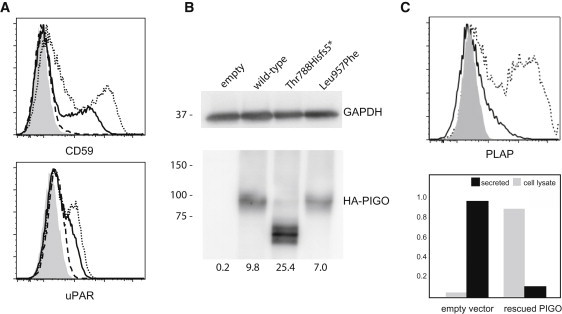
Hyperphosphatasia with mental retardation syndrome (HPMRS), an autosomal-recessive form of intellectual disability characterized by facial dysmorphism, seizures, brachytelephalangy, and persistent elevated serum alkaline phosphatase (hyperphosphatasia), was recently shown to be caused by mutations in PIGV, a member of the glycosylphosphatidylinositol (GPI)-anchor-synthesis pathway. However, not all individuals with HPMRS harbor mutations in this gene. By exome sequencing, we detected compound-heterozygous mutations in PIGO, a gene coding for a membrane protein of the same molecular pathway, in two siblings with HPMRS, and we then found by Sanger sequencing further mutations in another affected individual; these mutations cosegregated in the investigated families. The mutant transcripts are aberrantly spliced, decrease the membrane stability of the protein, or impair enzyme function such that GPI-anchor synthesis is affected and the level of GPI-anchored substrates localized at the cell surface is reduced. Our data identify PIGO as the second gene associated with HPMRS and suggest that a deficiency in GPI-anchor synthesis is the underlying molecular pathomechanism of HPMRS.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Kinoshita T., Fujita M., Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: Recent progress. J. Biochem. 2008;144:287–294. - PubMed
-
- Jaeken J. Congenital disorders of glycosylation (CDG): It's (nearly) all in it! J. Inherit. Metab. Dis. 2011;34:853–858. - PubMed
-
- Takeda J., Miyata T., Kawagoe K., Iida Y., Endo Y., Fujita T., Takahashi M., Kitani T., Kinoshita T. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
