

TMEM165 deficiency causes a congenital disorder of glycosylation
- PMID: 22683087
- PMCID: PMC3397274
- DOI: 10.1016/j.ajhg.2012.05.002
TMEM165 deficiency causes a congenital disorder of glycosylation
Abstract
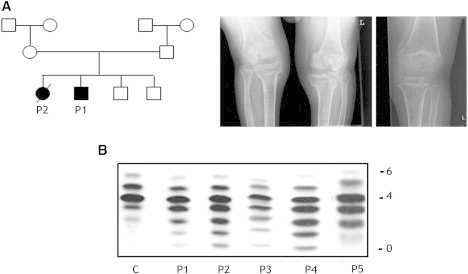
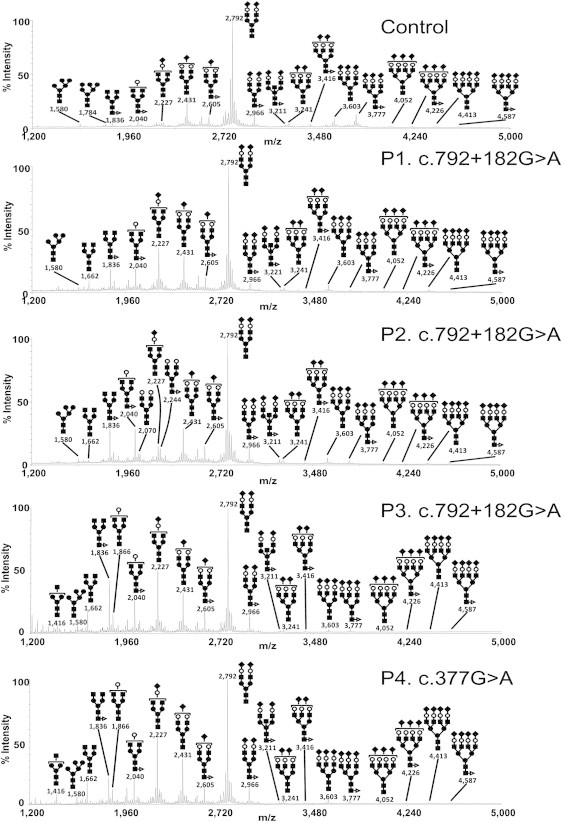
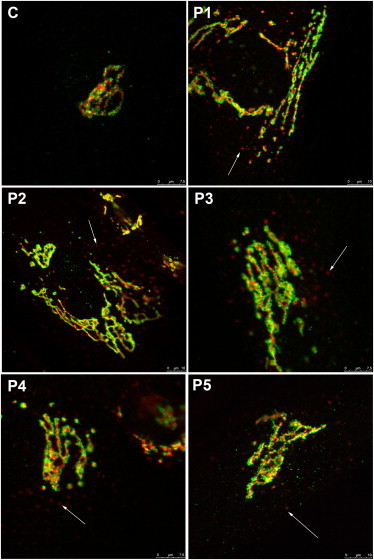
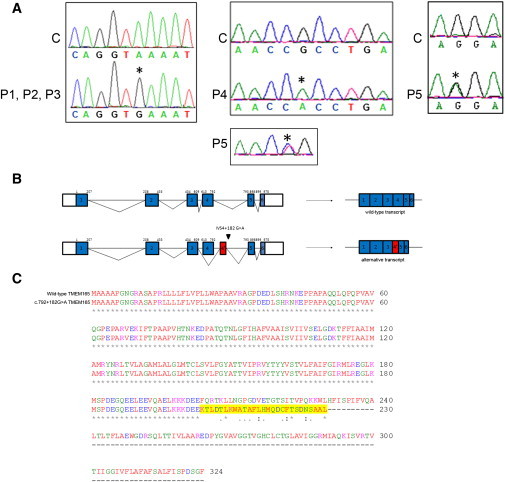
Protein glycosylation is a complex process that depends not only on the activities of several enzymes and transporters but also on a subtle balance between vesicular Golgi trafficking, compartmental pH, and ion homeostasis. Through a combination of autozygosity mapping and expression analysis in two siblings with an abnormal serum-transferrin isoelectric focusing test (type 2) and a peculiar skeletal phenotype with epiphyseal, metaphyseal, and diaphyseal dysplasia, we identified TMEM165 (also named TPARL) as a gene involved in congenital disorders of glycosylation (CDG). The affected individuals are homozygous for a deep intronic splice mutation in TMEM165. In our cohort of unsolved CDG-II cases, we found another individual with the same mutation and two unrelated individuals with missense mutations in TMEM165. TMEM165 encodes a putative transmembrane 324 amino acid protein whose cellular functions are unknown. Using a siRNA strategy, we showed that TMEM165 deficiency causes Golgi glycosylation defects in HEK cells.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
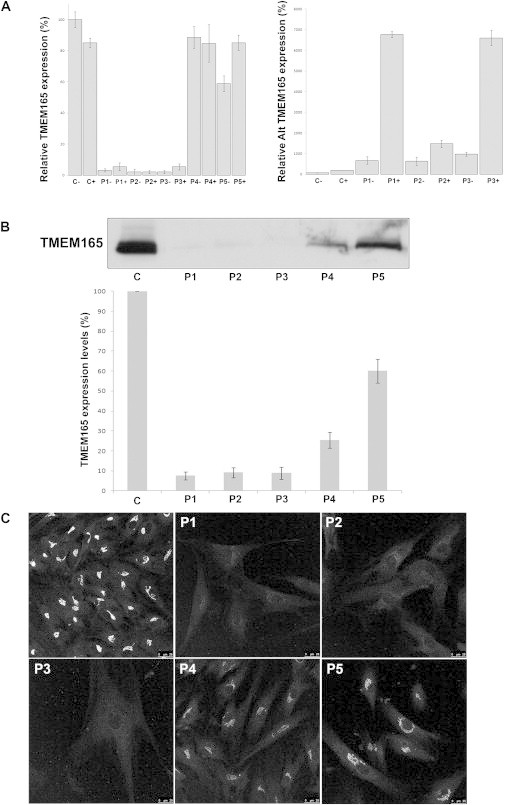
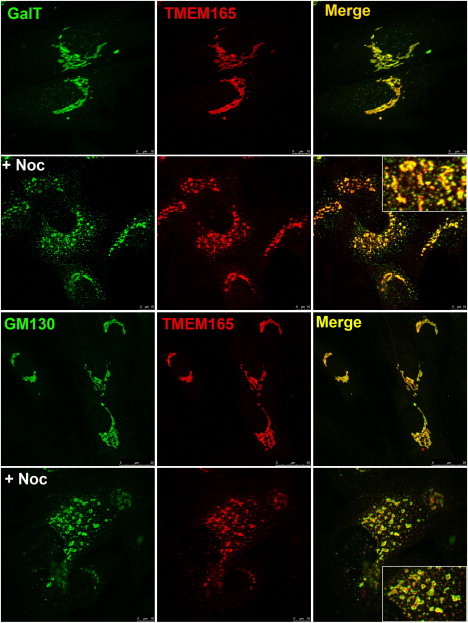
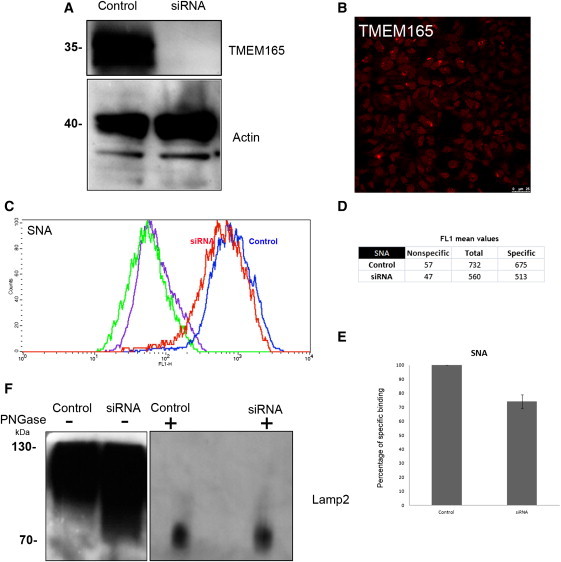
References
-
- Jaeken J., Matthijs G. Congenital disorders of glycosylation: A rapidly expanding disease family. Annu. Rev. Genomics Hum. Genet. 2007;8:261–278. - PubMed
-
- Foulquier F. COG defects, birth and rise! Biochim. Biophys. Acta. 2009;1792:896–902. - PubMed
-
- Wu X., Steet R.A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., Freeze H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004;10:518–523. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
