

Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants
- PMID: 22683818
- PMCID: PMC3432152
- DOI: 10.1016/j.freeradbiomed.2012.05.036
Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants
Abstract
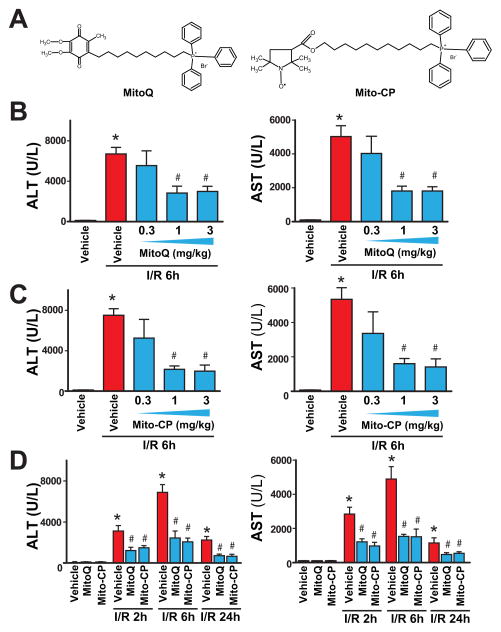
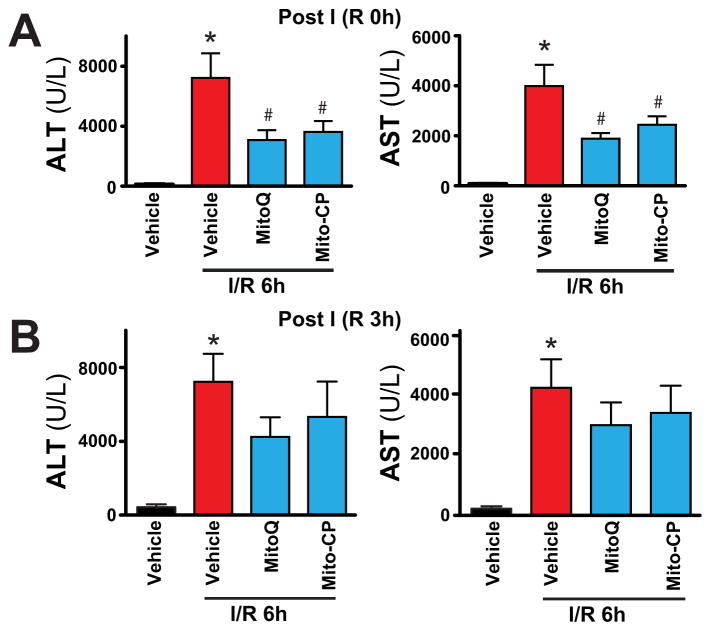
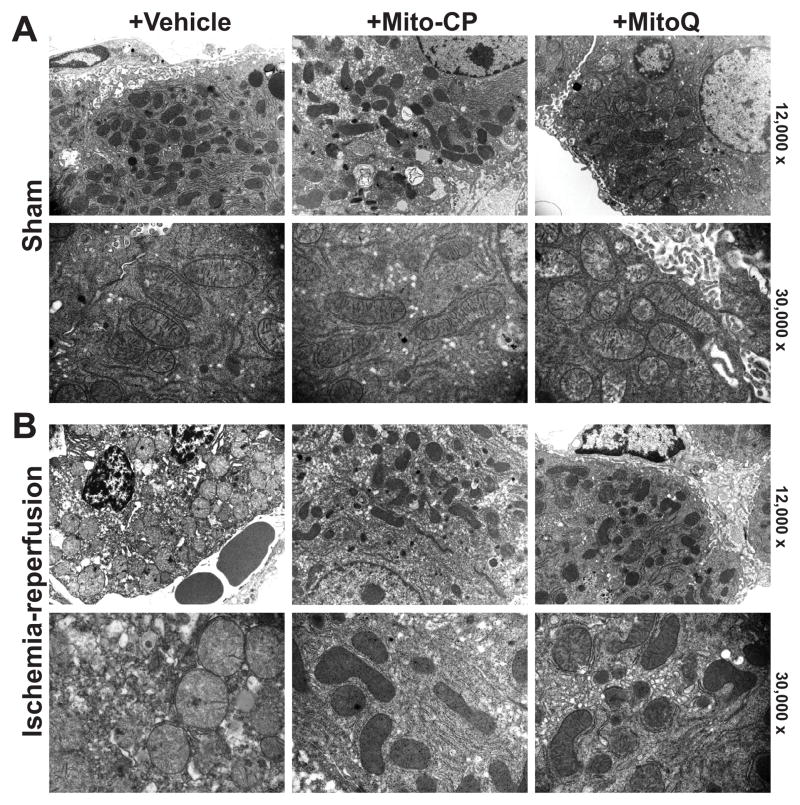
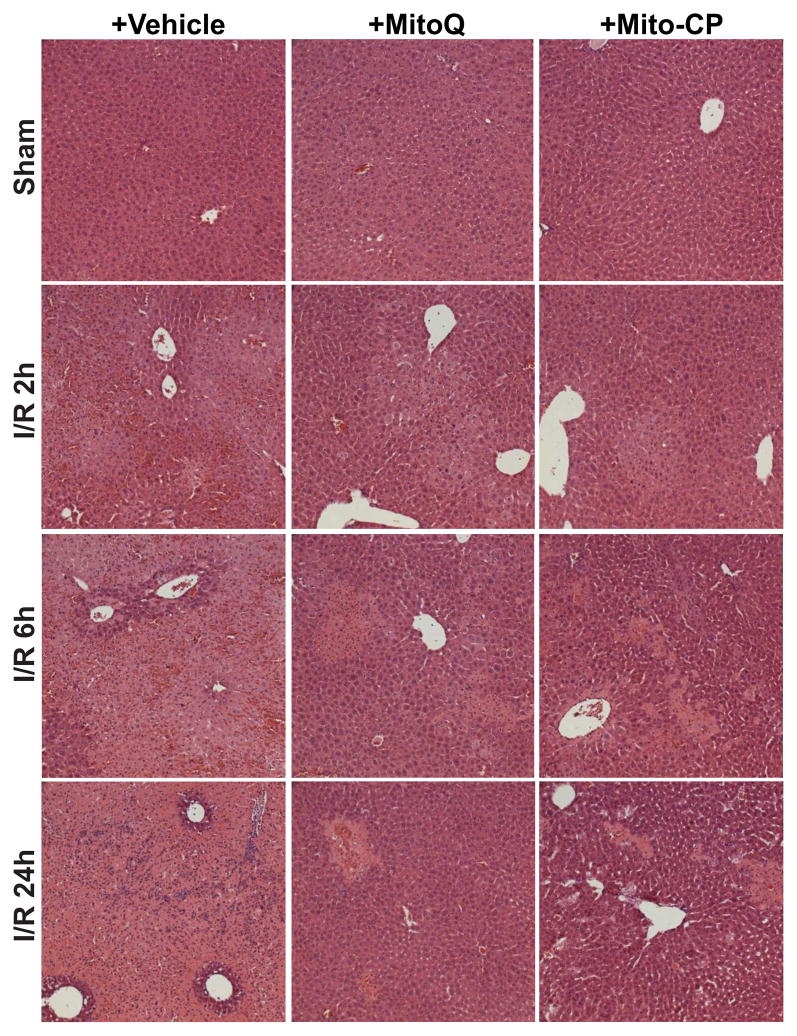
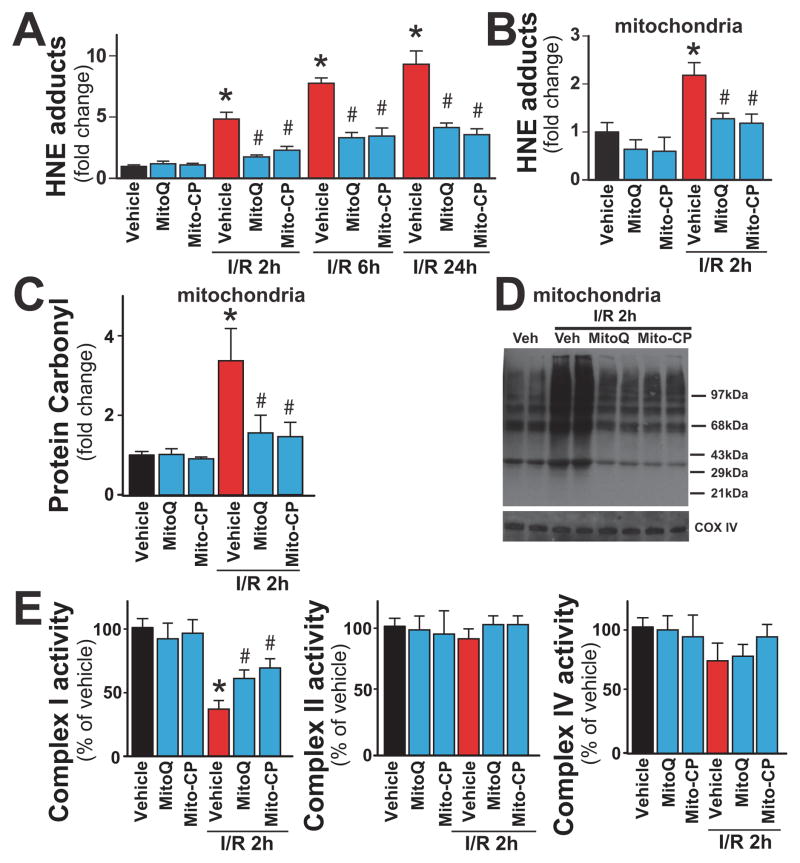
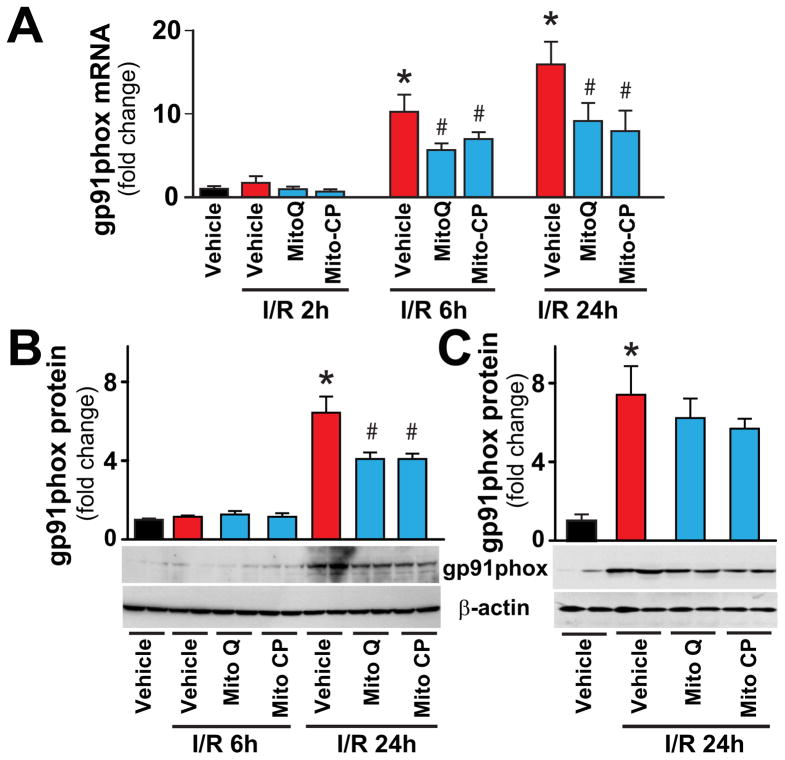
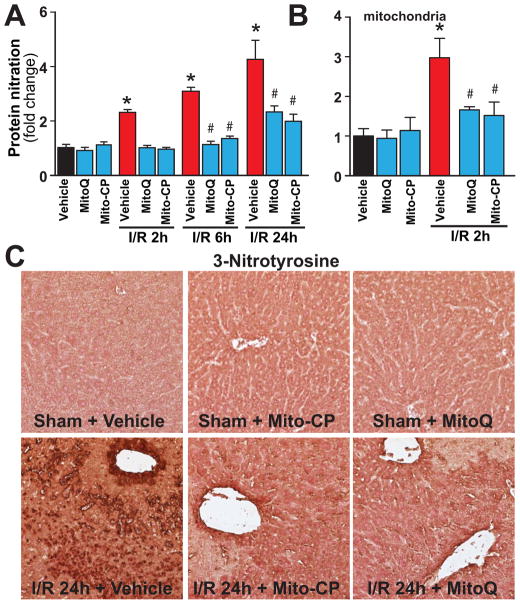
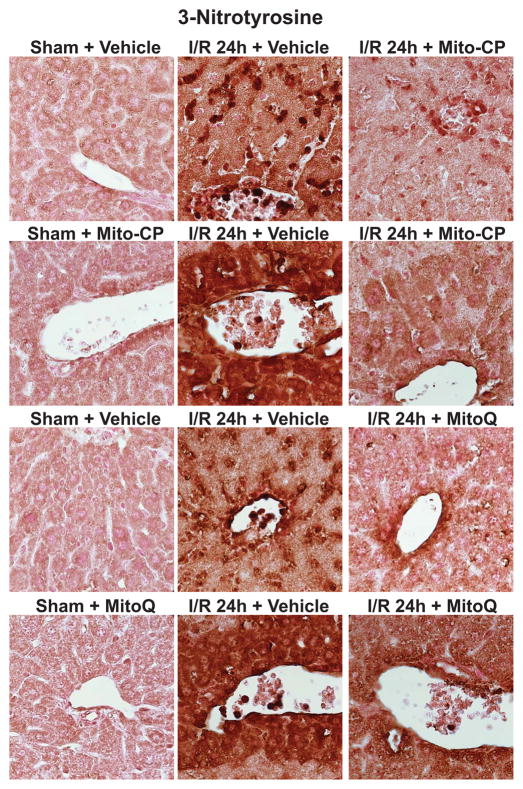
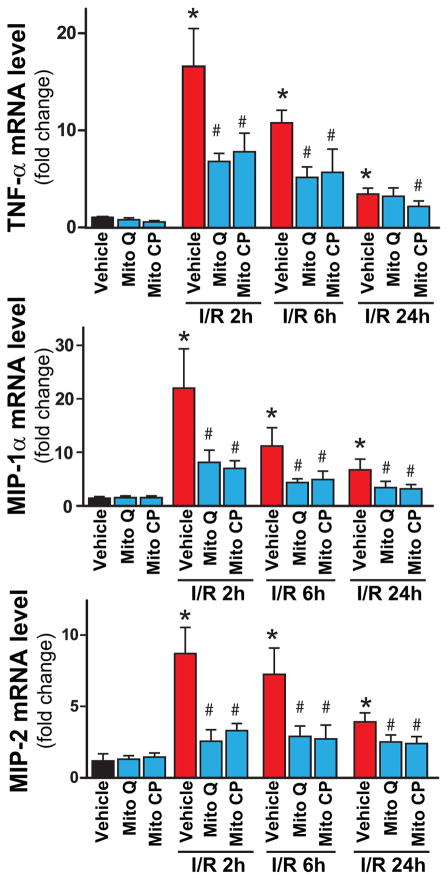
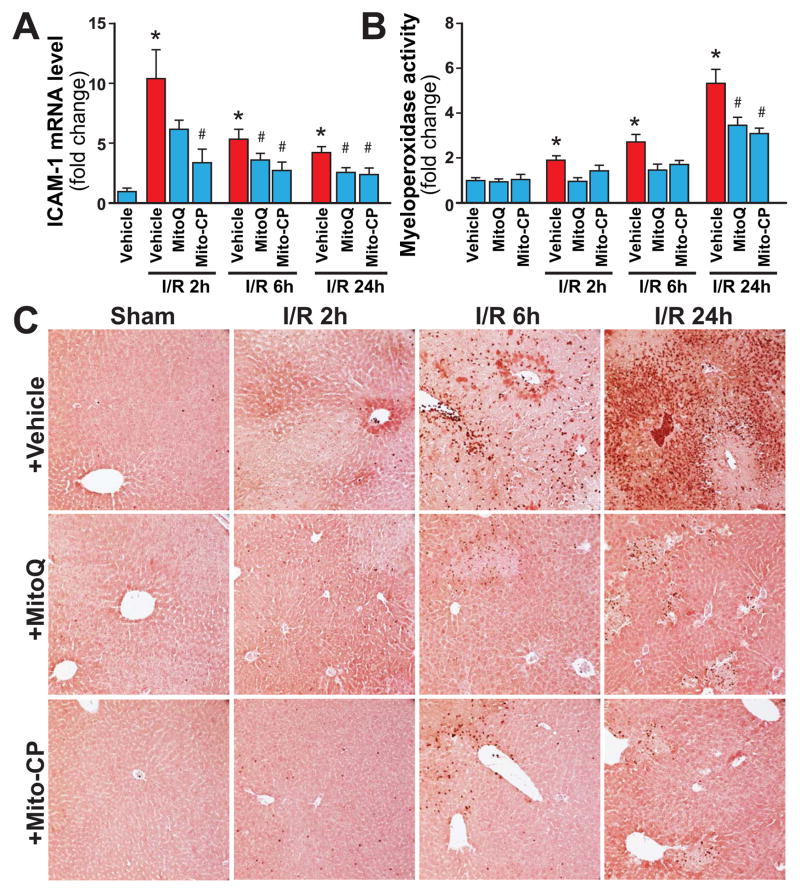
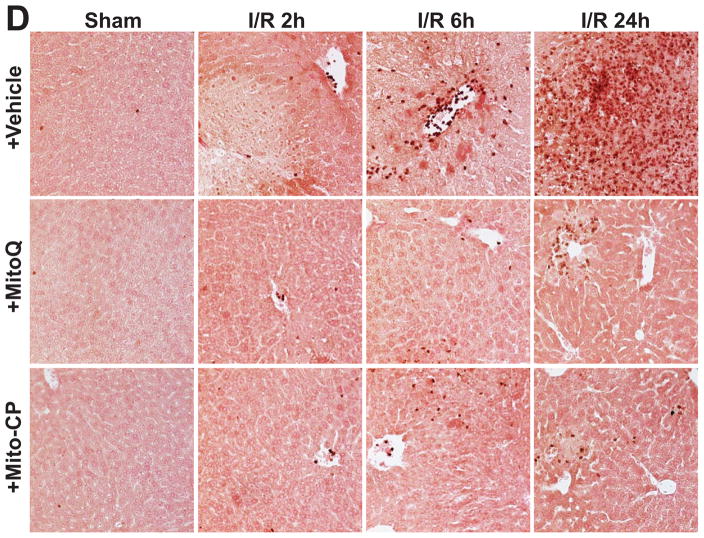
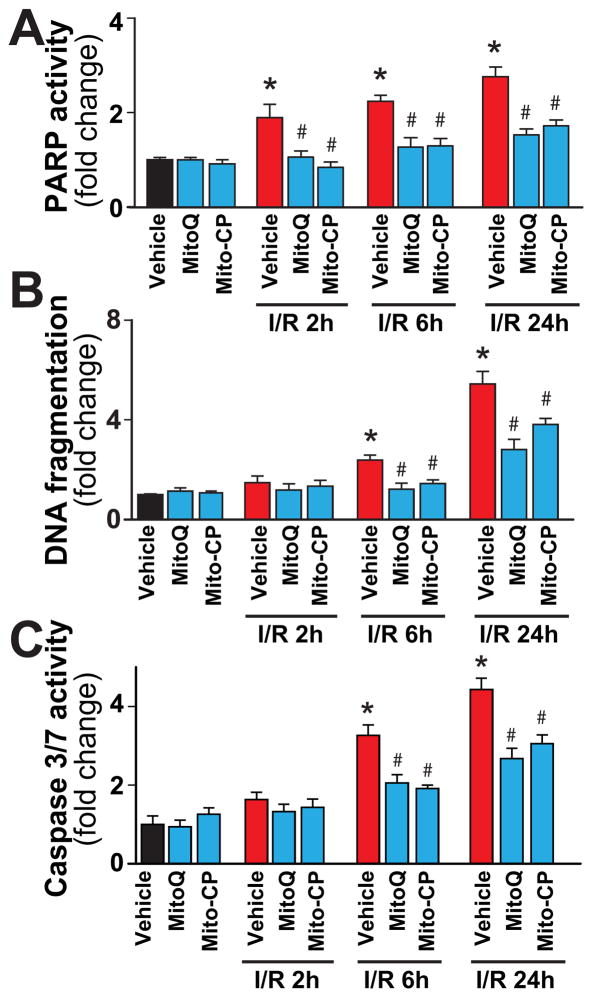
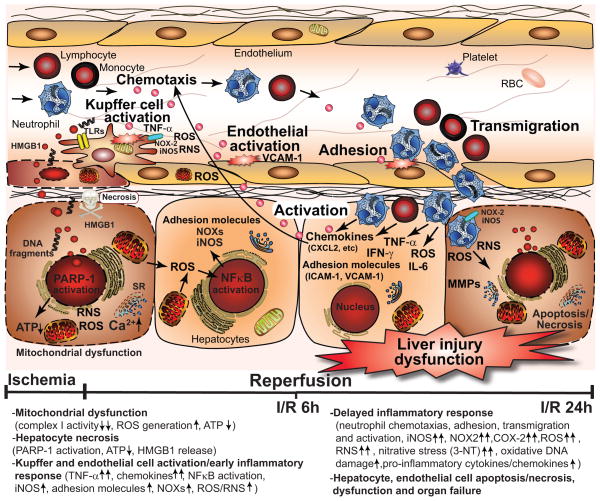
Mitochondrial reactive oxygen species generation has been implicated in the pathophysiology of ischemia-reperfusion (I/R) injury; however, its exact role and its spatial-temporal relationship with inflammation are elusive. Herein we explore the spatial-temporal relationship of oxidative/nitrative stress and inflammatory response during the course of hepatic I/R and the possible therapeutic potential of mitochondrial-targeted antioxidants, using a mouse model of segmental hepatic ischemia-reperfusion injury. Hepatic I/R was characterized by early (at 2 h of reperfusion) mitochondrial injury, decreased complex I activity, increased oxidant generation in the liver or liver mitochondria, and profound hepatocellular injury/dysfunction with acute proinflammatory response (TNF-α, MIP-1α/CCL3, MIP-2/CXCL2) without inflammatory cell infiltration, followed by marked neutrophil infiltration and a more pronounced secondary wave of oxidative/nitrative stress in the liver (starting from 6 h of reperfusion and peaking at 24 h). Mitochondrially targeted antioxidants, MitoQ or Mito-CP, dose-dependently attenuated I/R-induced liver dysfunction, the early and delayed oxidative and nitrative stress response (HNE/carbonyl adducts, malondialdehyde, 8-OHdG, and 3-nitrotyrosine formation), and mitochondrial and histopathological injury/dysfunction, as well as delayed inflammatory cell infiltration and cell death. Mitochondrially generated oxidants play a central role in triggering the deleterious cascade of events associated with hepatic I/R, which may be targeted by novel antioxidants for therapeutic advantage.
Published by Elsevier Inc.
Figures

Similar articles
-
Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy.Free Radic Biol Med. 2012 Jan 15;52(2):497-506. doi: 10.1016/j.freeradbiomed.2011.11.001. Epub 2011 Nov 10. Free Radic Biol Med. 2012. PMID: 22120494 Free PMC article.
-
Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ.Redox Biol. 2015 Aug;5:163-168. doi: 10.1016/j.redox.2015.04.008. Epub 2015 Apr 29. Redox Biol. 2015. PMID: 25965144 Free PMC article.
-
The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model.J Heart Lung Transplant. 2015 Nov;34(11):1471-80. doi: 10.1016/j.healun.2015.05.007. Epub 2015 Jun 11. J Heart Lung Transplant. 2015. PMID: 26140808 Free PMC article.
-
Oxidative stress in hepatic ischemia-reperfusion injury: the role of antioxidants and iron chelating compounds.Curr Pharm Des. 2006;12(23):2875-90. doi: 10.2174/138161206777947614. Curr Pharm Des. 2006. PMID: 16918418 Review.
-
Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers.Int J Surg. 2016 Sep;33 Suppl 1:S57-70. doi: 10.1016/j.ijsu.2016.05.050. Epub 2016 May 30. Int J Surg. 2016. PMID: 27255130
Cited by
-
PGC-1α Protects against Hepatic Ischemia Reperfusion Injury by Activating PPARα and PPARγ and Regulating ROS Production.Oxid Med Cell Longev. 2021 May 19;2021:6677955. doi: 10.1155/2021/6677955. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34104311 Free PMC article.
-
Nicotinamide mononucleotide preserves mitochondrial function and increases survival in hemorrhagic shock.JCI Insight. 2018 Sep 6;3(17):e120182. doi: 10.1172/jci.insight.120182. eCollection 2018 Sep 6. JCI Insight. 2018. PMID: 30185676 Free PMC article.
-
Solvent- and Catalyst-Free Environmentally Benign High Hydrostatic Pressure-Assisted Synthesis of Bioactive Hydrazones and the Evaluation of Their Stability Under Various Storage Conditions.Molecules. 2024 Nov 8;29(22):5287. doi: 10.3390/molecules29225287. Molecules. 2024. PMID: 39598676 Free PMC article.
-
Mitochondria-Targeted Antioxidants: Future Perspectives in Kidney Ischemia Reperfusion Injury.Oxid Med Cell Longev. 2016;2016:2950503. doi: 10.1155/2016/2950503. Epub 2016 May 24. Oxid Med Cell Longev. 2016. PMID: 27313826 Free PMC article. Review.
-
Advances in therapeutic options for portal hypertension.Therap Adv Gastroenterol. 2018 Nov 25;11:1756284818811294. doi: 10.1177/1756284818811294. eCollection 2018. Therap Adv Gastroenterol. 2018. PMID: 30505350 Free PMC article. Review.
References
-
- Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 2003;284:G15–26. - PubMed
-
- Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: Present concepts. J Gastroenterol Hepatol. 2011;26(Suppl 1):173–179. - PubMed
-
- Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. - PubMed
-
- Szabo C. The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia-reperfusion injury. Shock. 1996;6:79–88. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
