

Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production
- PMID: 22684298
- PMCID: PMC3383590
- DOI: 10.4161/cc.20718
Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production
Abstract
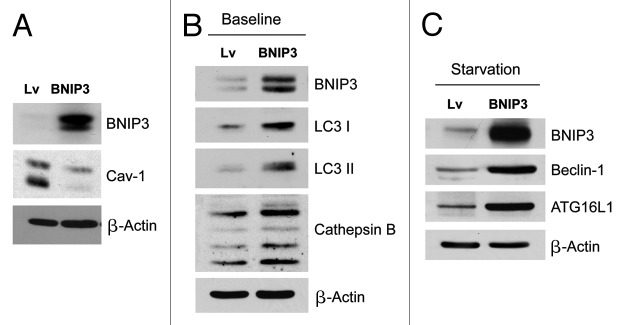
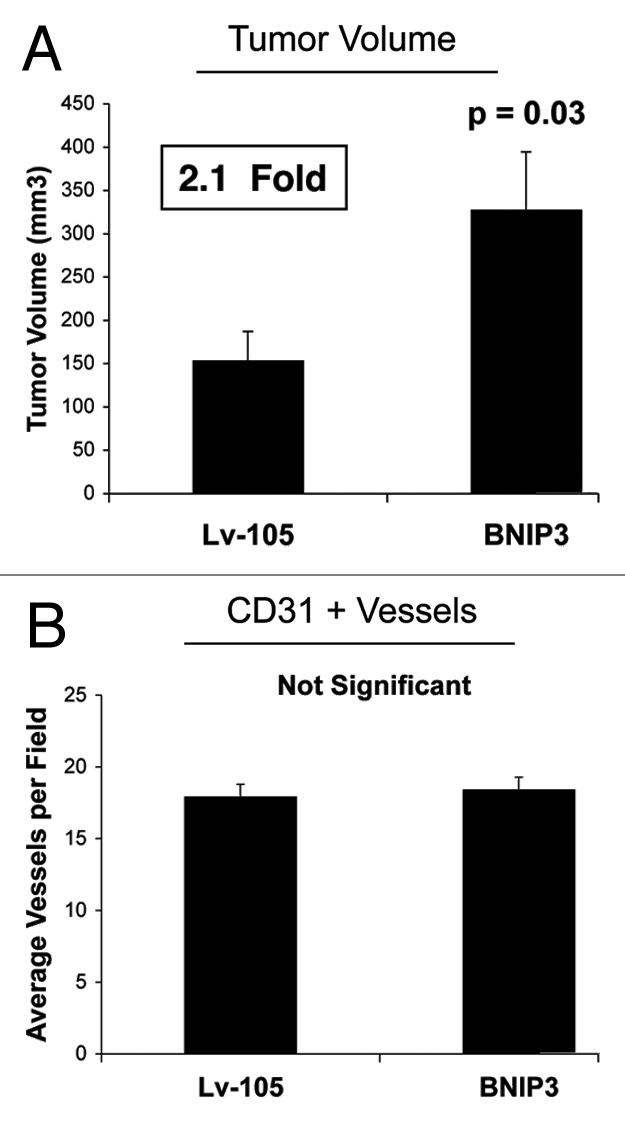
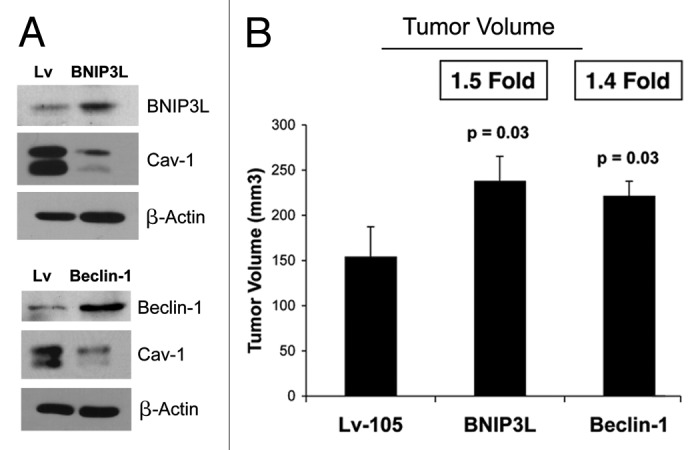
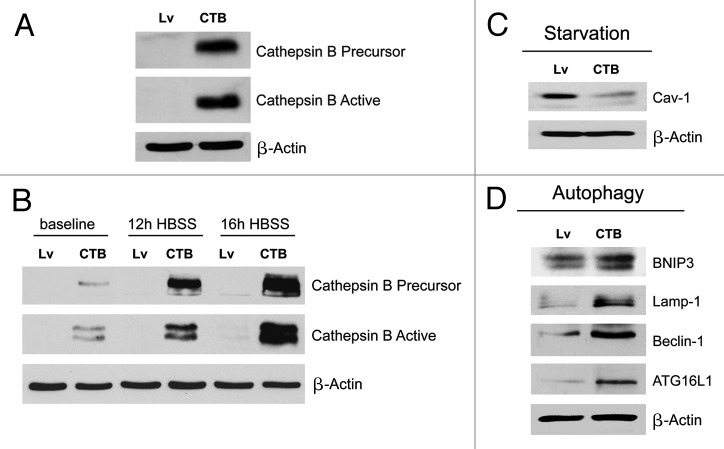
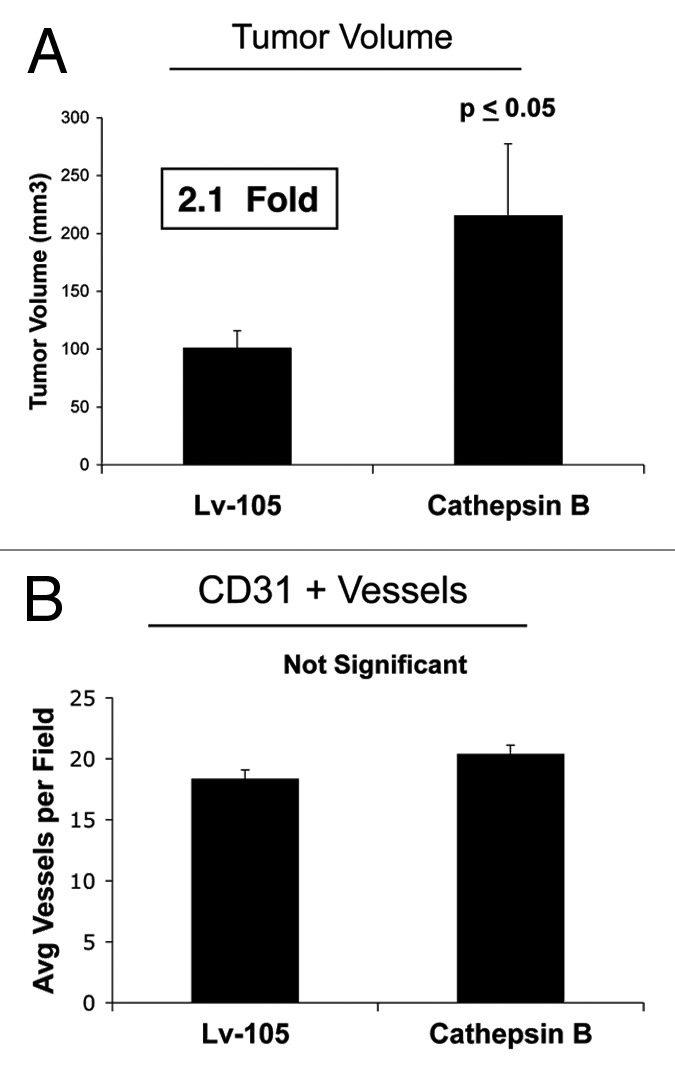
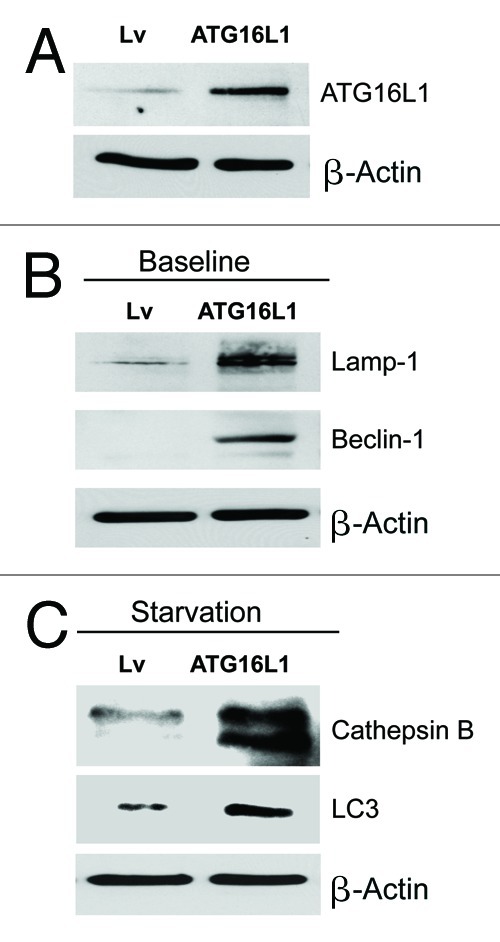
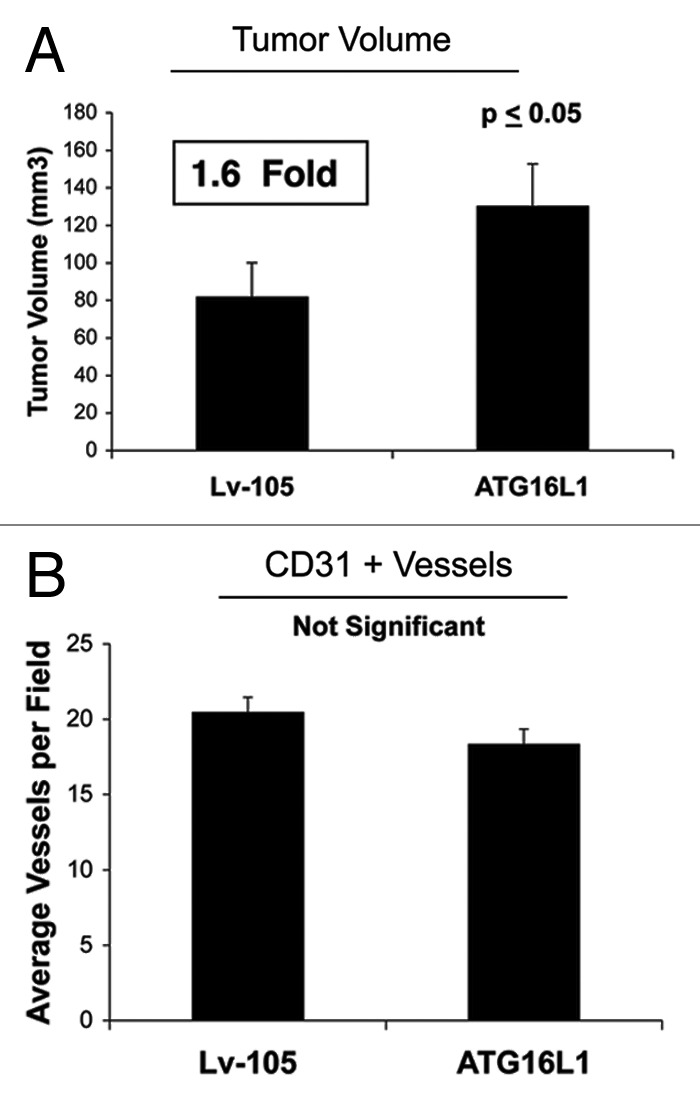
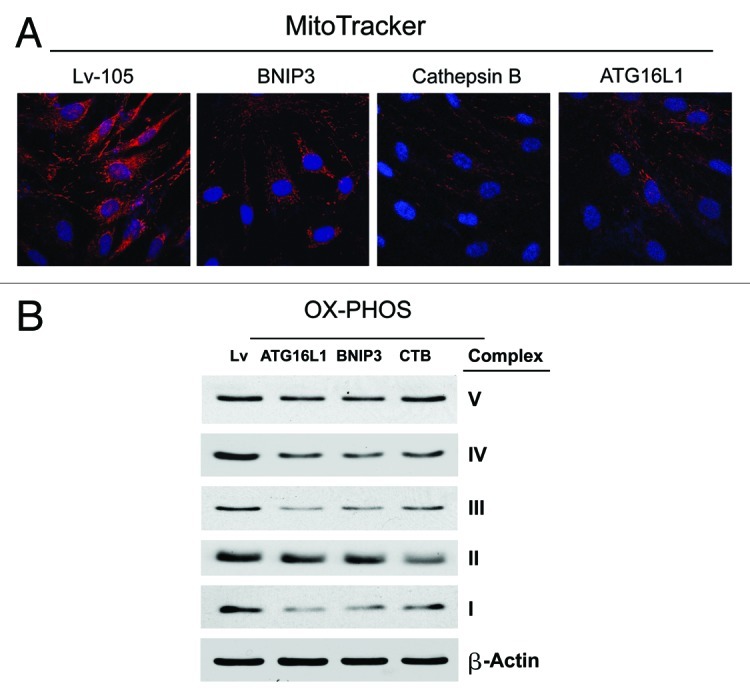
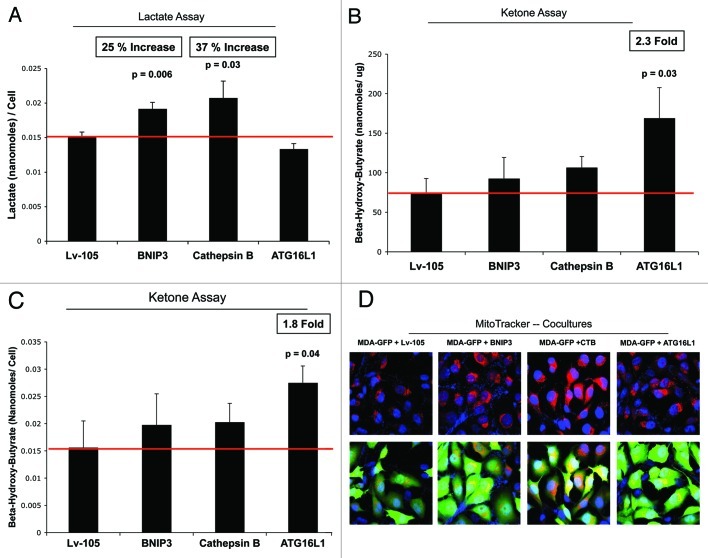
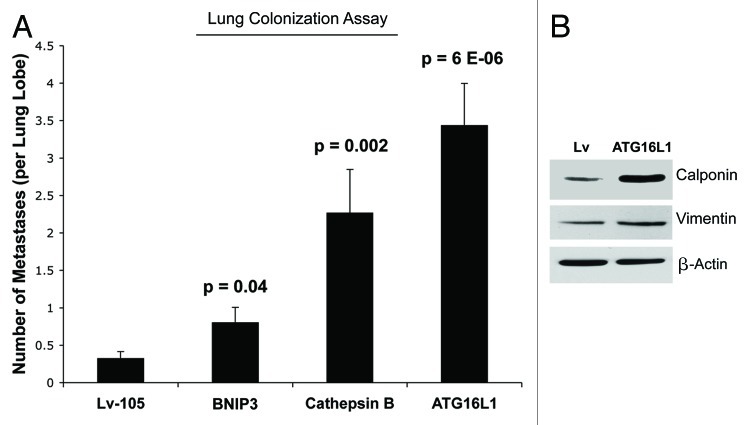
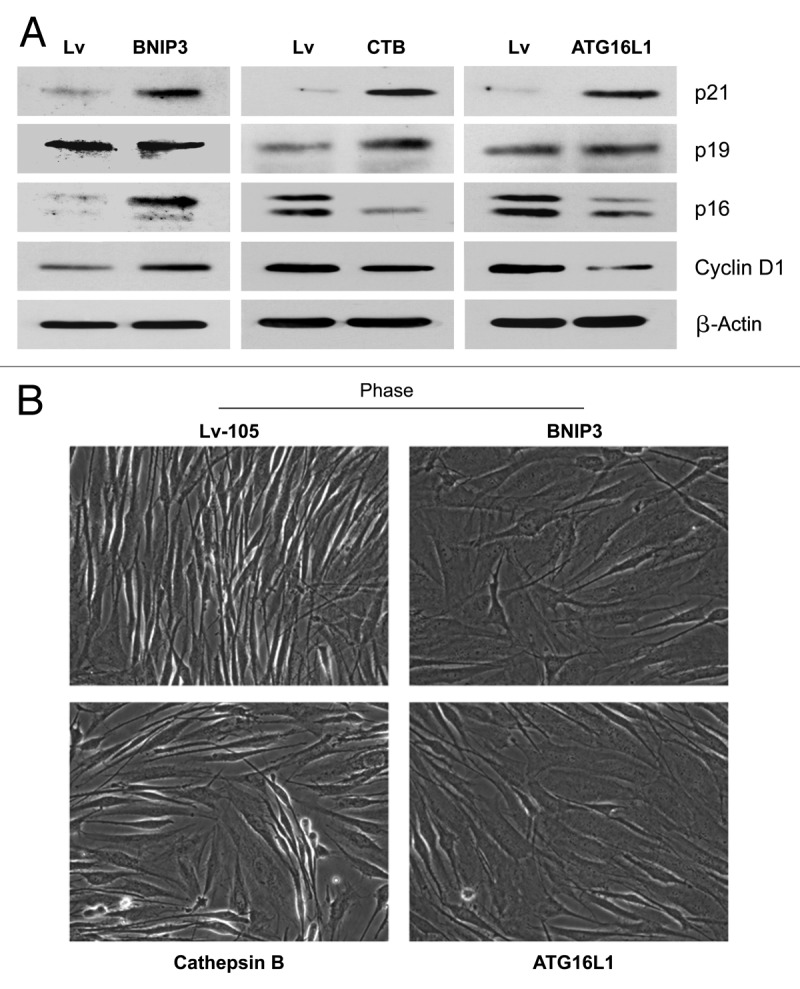
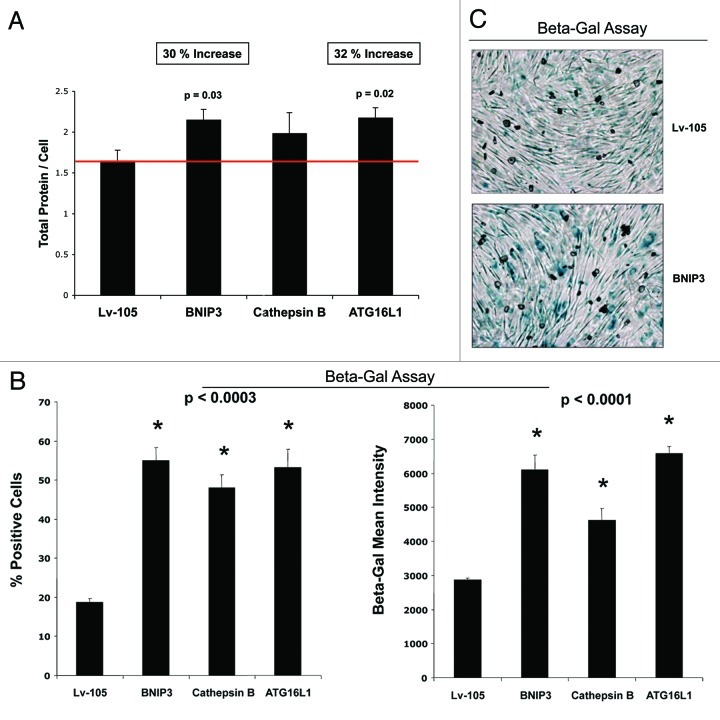
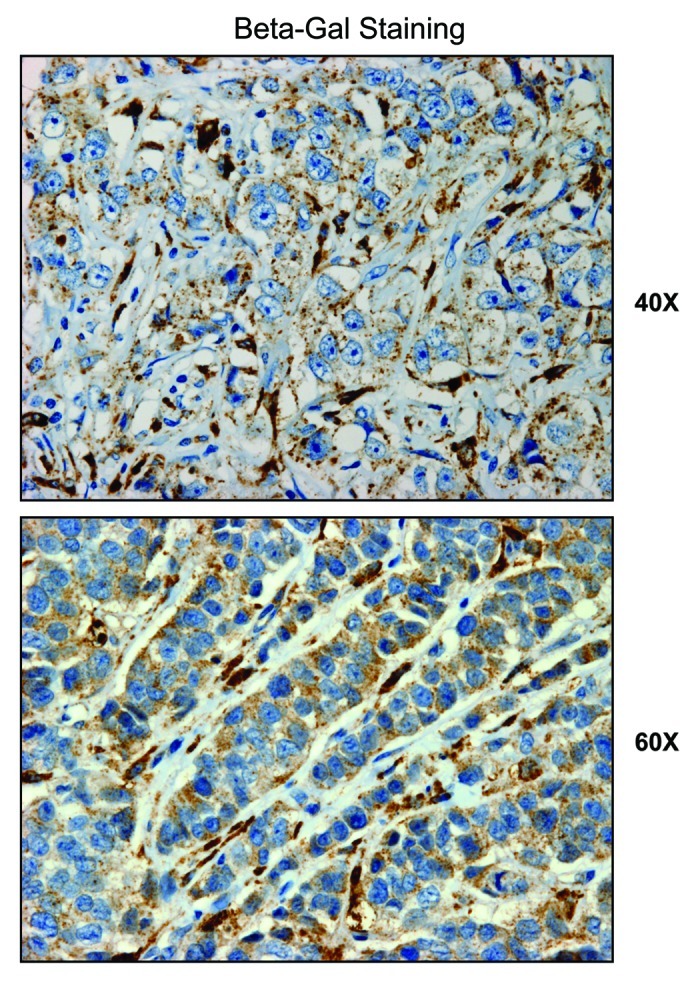
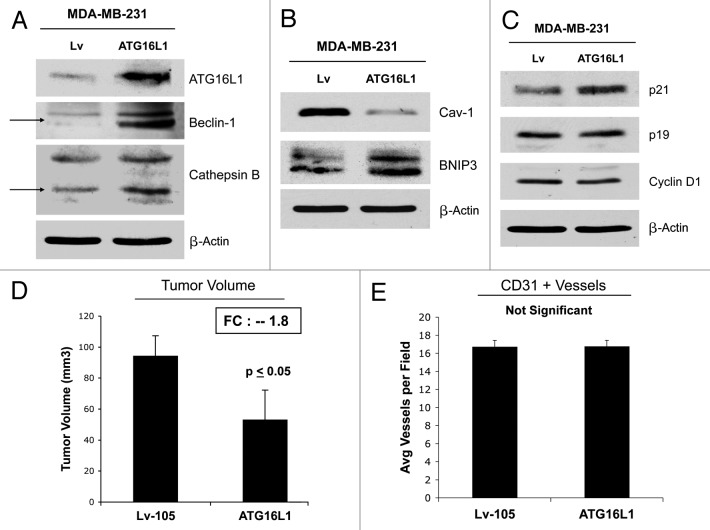
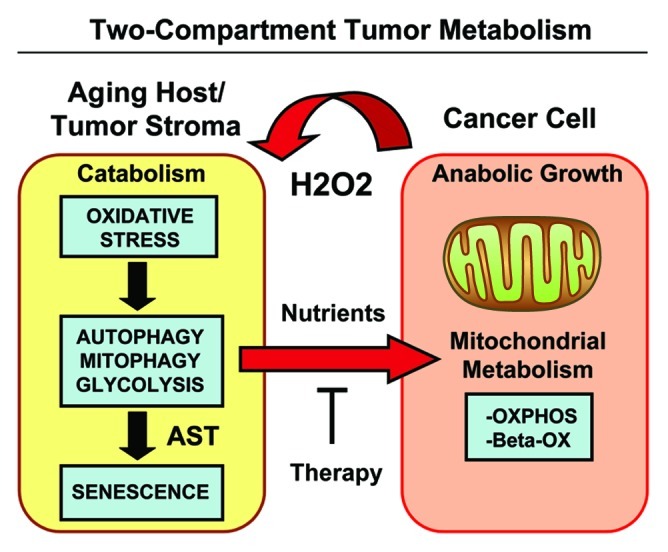
Senescent fibroblasts are known to promote tumor growth. However, the exact mechanism remains largely unknown. An important clue comes from recent studies linking autophagy with the onset of senescence. Thus, autophagy and senescence may be part of the same physiological process, known as the autophagy-senescence transition (AST). To test this hypothesis, human fibroblasts immortalized with telomerase (hTERT-BJ1) were stably transfected with autophagy genes (BNIP3, CTSB or ATG16L1). Their overexpression was sufficient to induce a constitutive autophagic phenotype, with features of mitophagy, mitochondrial dysfunction and a shift toward aerobic glycolysis, resulting in L-lactate and ketone body production. Autophagic fibroblasts also showed features of senescence, with increased p21(WAF1/CIP1), a CDK inhibitor, cellular hypertrophy and increased β-galactosidase activity. Thus, we genetically validated the existence of the autophagy-senescence transition. Importantly, autophagic-senescent fibroblasts promoted tumor growth and metastasis, when co-injected with human breast cancer cells, independently of angiogenesis. Autophagic-senescent fibroblasts stimulated mitochondrial metabolism in adjacent cancer cells, when the two cell types were co-cultured, as visualized by MitoTracker staining. In particular, autophagic ATG16L1 fibroblasts, which produced large amounts of ketone bodies (3-hydroxy-butyrate), had the strongest effects and promoted metastasis by up to 11-fold. Conversely, expression of ATG16L1 in epithelial cancer cells inhibited tumor growth, indicating that the effects of autophagy are compartment-specific. Thus, autophagic-senescent fibroblasts metabolically promote tumor growth and metastasis, by paracrine production of high-energy mitochondrial fuels. Our current studies provide genetic support for the importance of "two-compartment tumor metabolism" in driving tumor growth and metastasis via a simple energy transfer mechanism. Finally, β-galactosidase, a known lysosomal enzyme and biomarker of senescence, was localized to the tumor stroma in human breast cancer tissues, providing in vivo support for our hypothesis. Bioinformatic analysis of genome-wide transcriptional profiles from tumor stroma, isolated from human breast cancers, also validated the onset of an autophagy-senescence transition. Taken together, these studies establish a new functional link between host aging, autophagy, the tumor microenvironment and cancer metabolism.
Figures
Comment in
-
Cancers co-opt cohabitants' catabolism: autophagy and senescence in the tumor stroma.Cell Cycle. 2012 Jun 15;11(12):2230-1. doi: 10.4161/cc.20964. Epub 2012 Jun 15. Cell Cycle. 2012. PMID: 22672908 Free PMC article.
-
CTGF-mediated autophagy-senescence transition in tumor stroma promotes anabolic tumor growth and metastasis.Cell Cycle. 2012 Jul 15;11(14):2592-3. doi: 10.4161/cc.21240. Epub 2012 Jul 15. Cell Cycle. 2012. PMID: 22751431 Free PMC article.
References
-
- Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, et al. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–63. doi: 10.4161/cc.10.13.16233. - DOI - PMC - PubMed
-
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer-associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA075503/CA/NCI NIH HHS/United States
- R01 CA098779/CA/NCI NIH HHS/United States
- R01-CA-120876/CA/NCI NIH HHS/United States
- R01 CA120876/CA/NCI NIH HHS/United States
- R01-CA-70896/CA/NCI NIH HHS/United States
- R01-CA-098779/CA/NCI NIH HHS/United States
- R01-CA-86072/CA/NCI NIH HHS/United States
- R01-AR-055660/AR/NIAMS NIH HHS/United States
- R01-CA-080250/CA/NCI NIH HHS/United States
- R01 CA070896/CA/NCI NIH HHS/United States
- R01 CA107382/CA/NCI NIH HHS/United States
- P30 CA056036/CA/NCI NIH HHS/United States
- P30-CA-56036/CA/NCI NIH HHS/United States
- R01-CA-107382/CA/NCI NIH HHS/United States
- R01 AR055660/AR/NIAMS NIH HHS/United States
- R01-CA-75503/CA/NCI NIH HHS/United States
- R01 CA080250/CA/NCI NIH HHS/United States
- R01 CA086072/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous