Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations
- PMID: 22685075
- PMCID: PMC3413386
- DOI: 10.1093/bioinformatics/bts336
Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations
Abstract
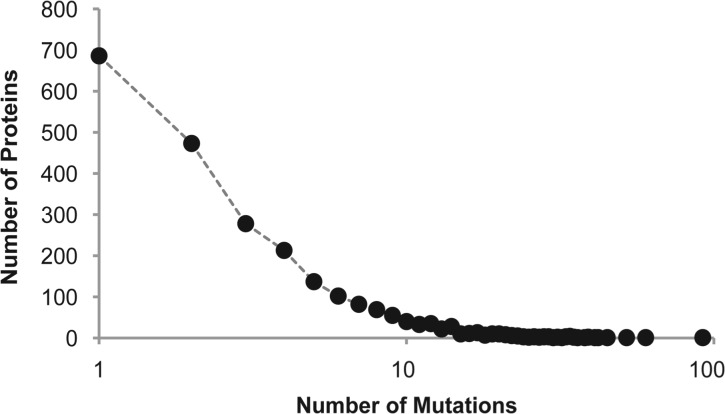
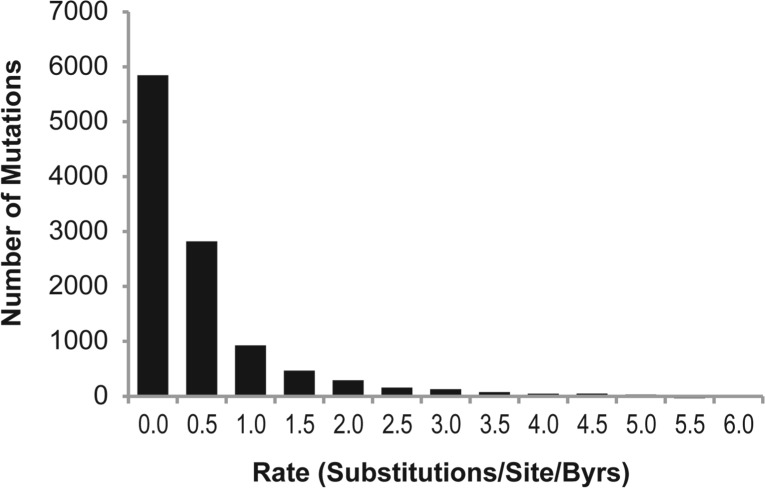
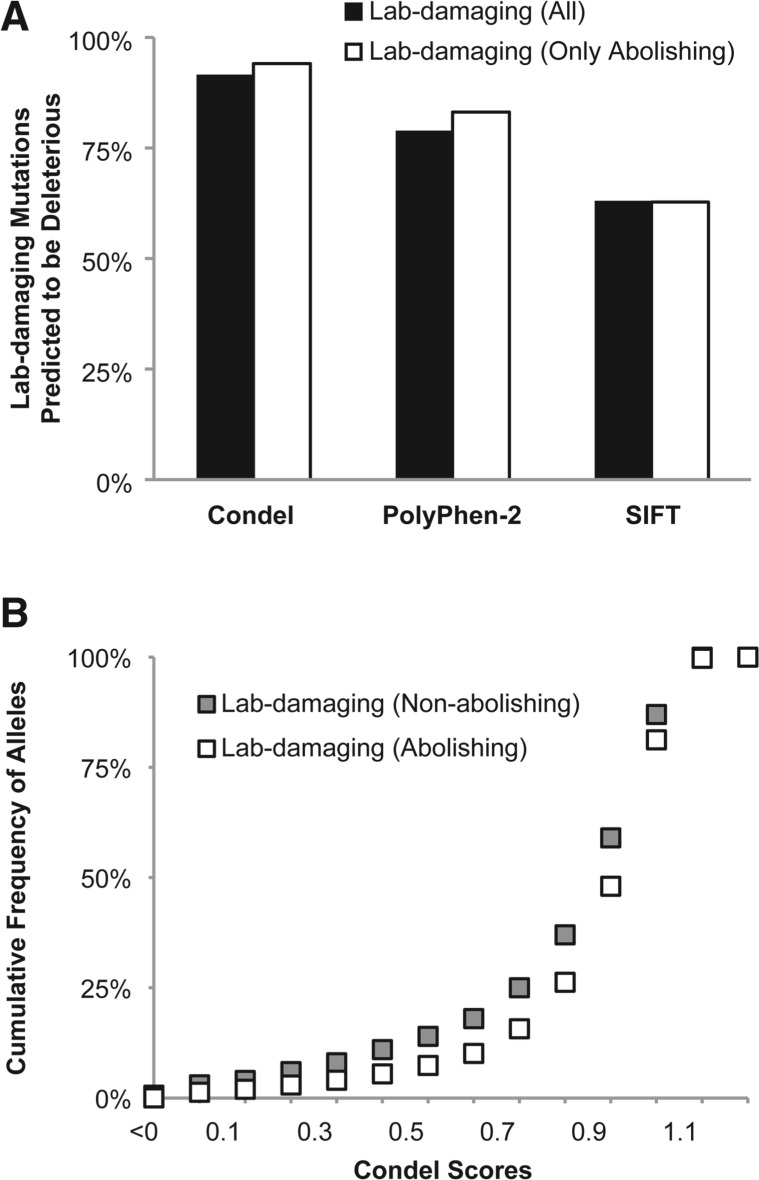
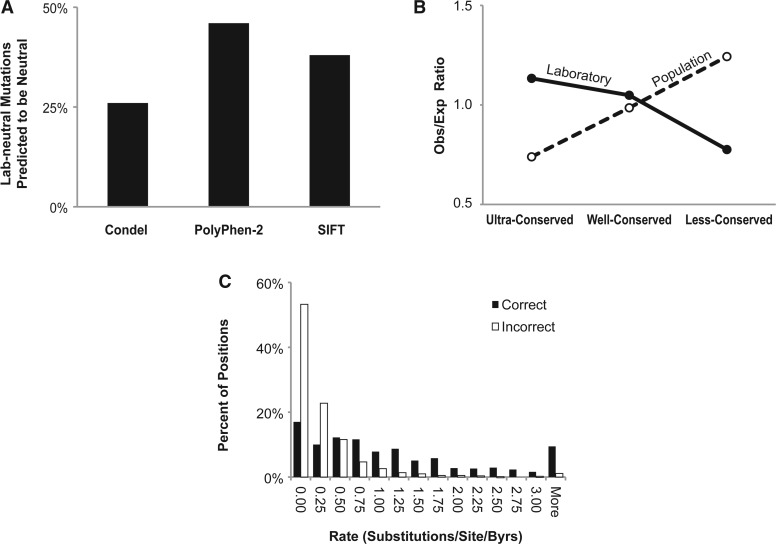
Site-directed mutagenesis is frequently used by scientists to investigate the functional impact of amino acid mutations in the laboratory. Over 10,000 such laboratory-induced mutations have been reported in the UniProt database along with the outcomes of functional assays. Here, we explore the performance of state-of-the-art computational tools (Condel, PolyPhen-2 and SIFT) in correctly annotating the function-altering potential of 10,913 laboratory-induced mutations from 2372 proteins. We find that computational tools are very successful in diagnosing laboratory-induced mutations that elicit significant functional change in the laboratory (up to 92% accuracy). But, these tools consistently fail in correctly annotating laboratory-induced mutations that show no functional impact in the laboratory assays. Therefore, the overall accuracy of computational tools for laboratory-induced mutations is much lower than that observed for the naturally occurring human variants. We tested and rejected the possibilities that the preponderance of changes to alanine and the presence of multiple base-pair mutations in the laboratory were the reasons for the observed discordance between the performance of computational tools for natural and laboratory mutations. Instead, we discover that the laboratory-induced mutations occur predominately at the highly conserved positions in proteins, where the computational tools have the lowest accuracy of correct prediction for variants that do not impact function (neutral). Therefore, the comparisons of experimental-profiling results with those from computational predictions need to be sensitive to the evolutionary conservation of the positions harboring the amino acid change.
Figures