

Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders
- PMID: 22685291
- PMCID: PMC3411061
- DOI: 10.1074/jbc.M112.346932
Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders
Abstract
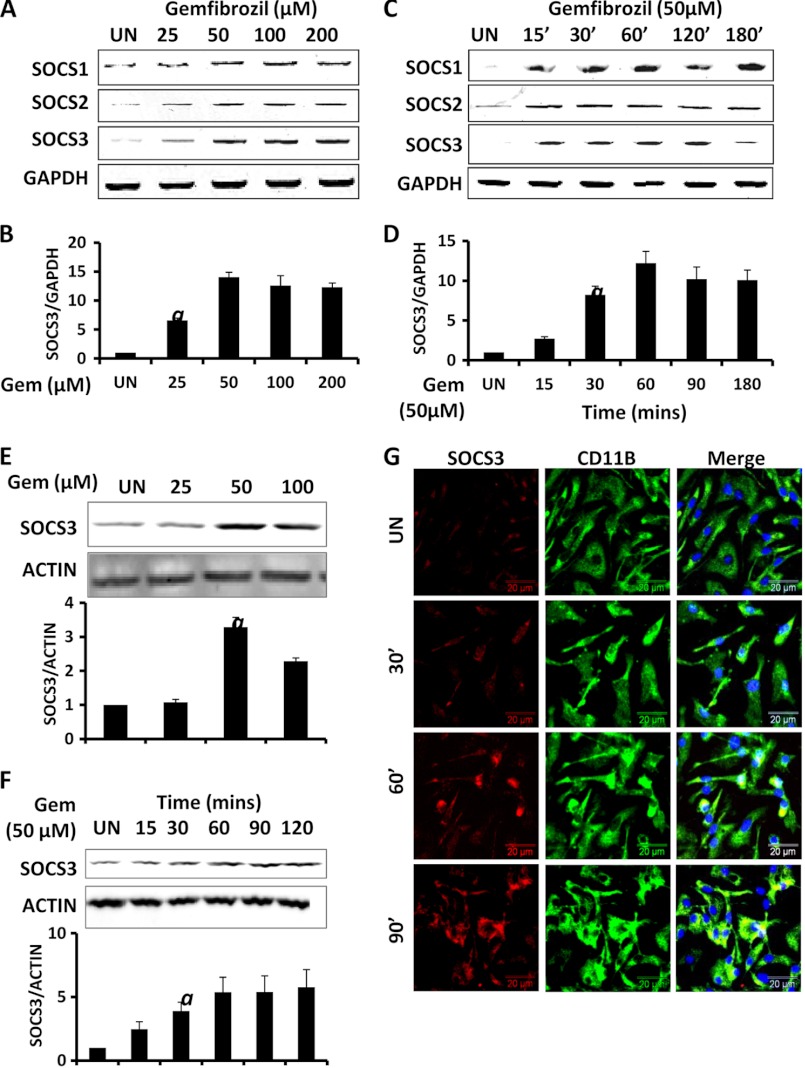
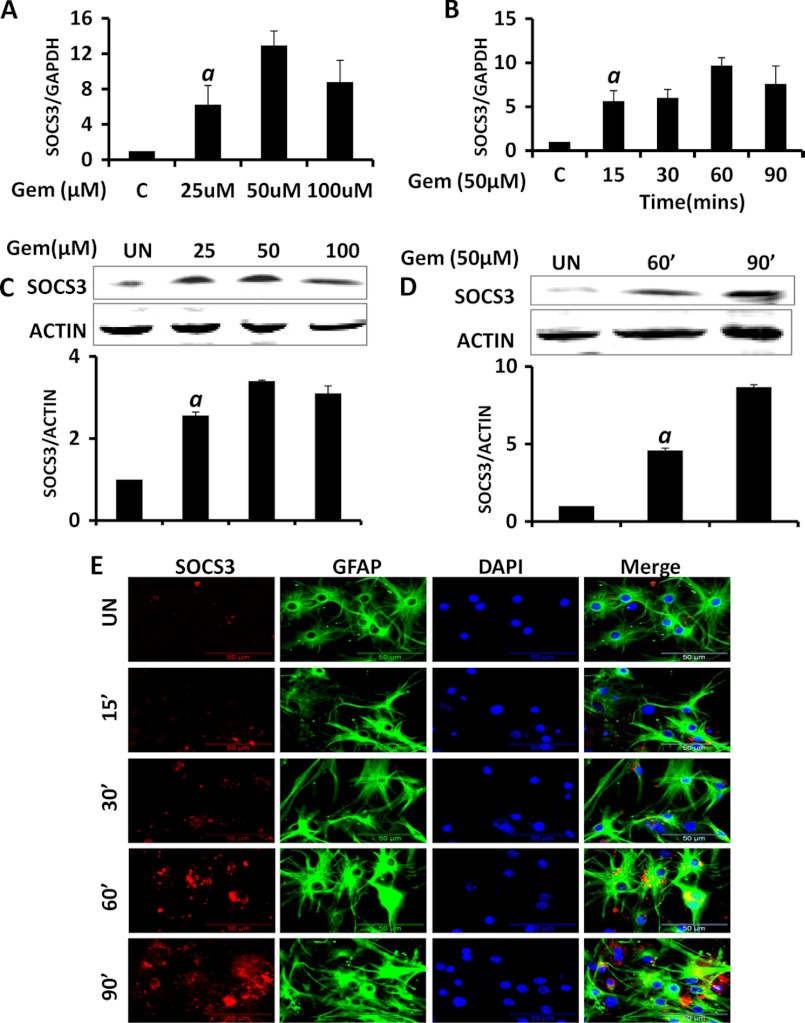
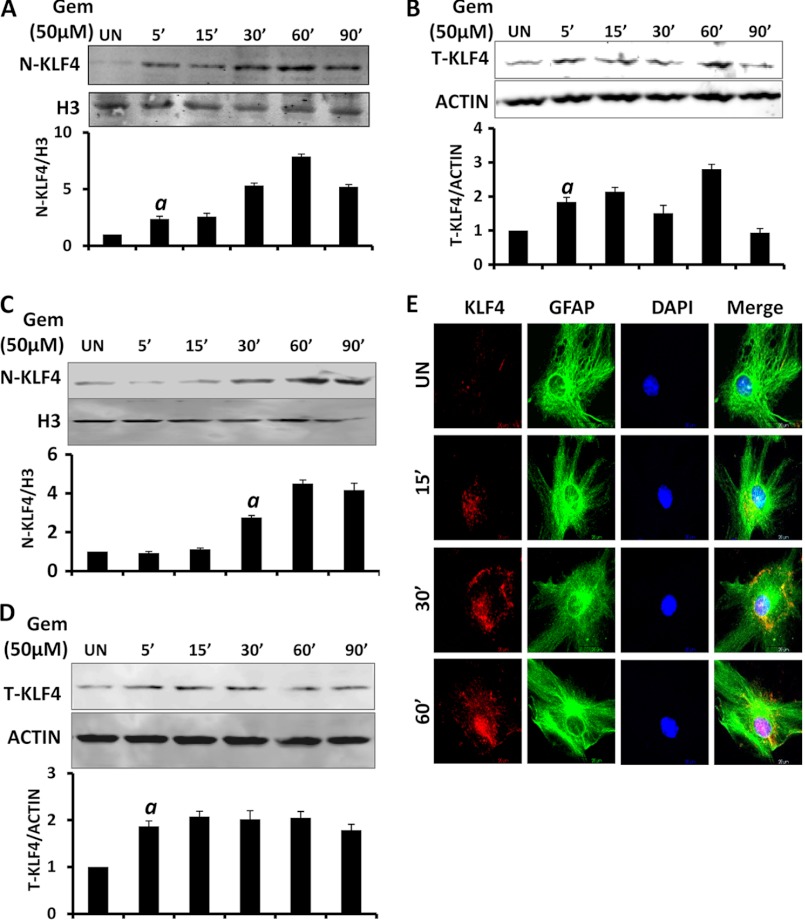
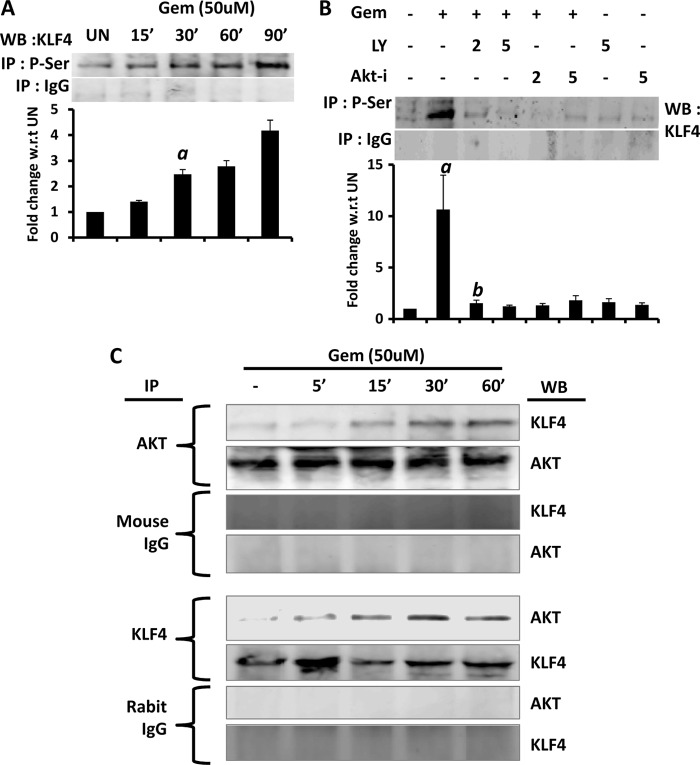
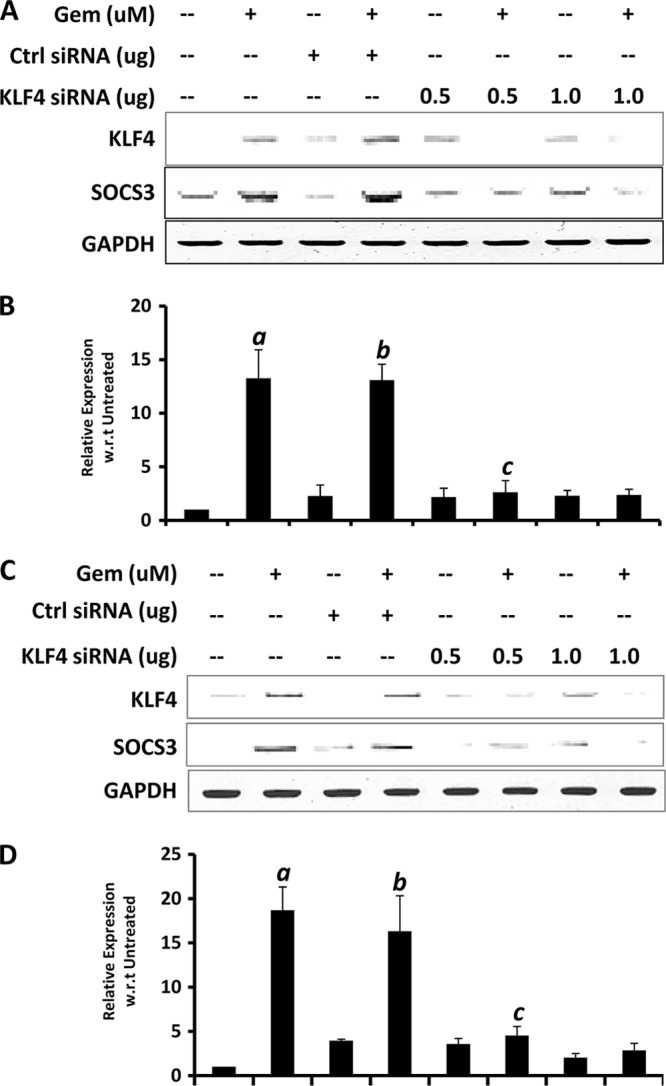
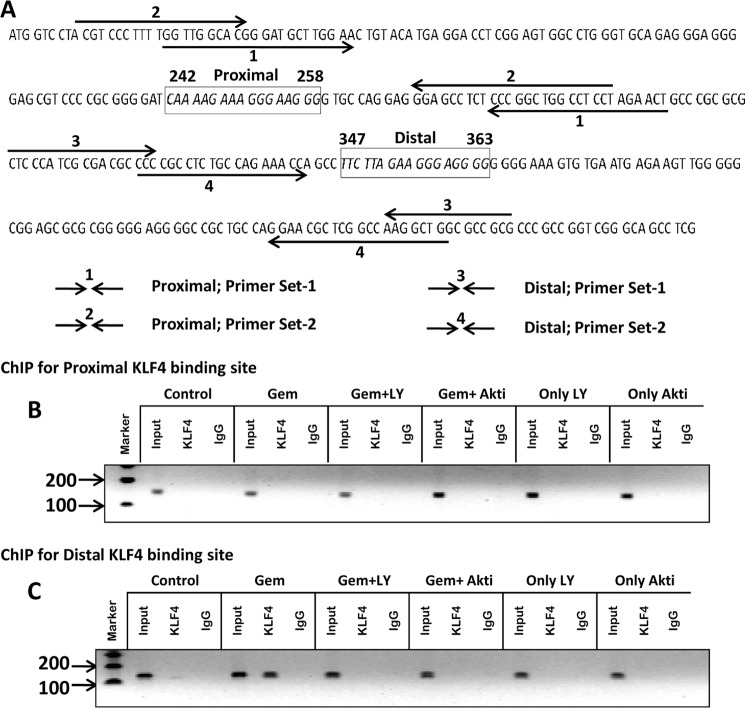
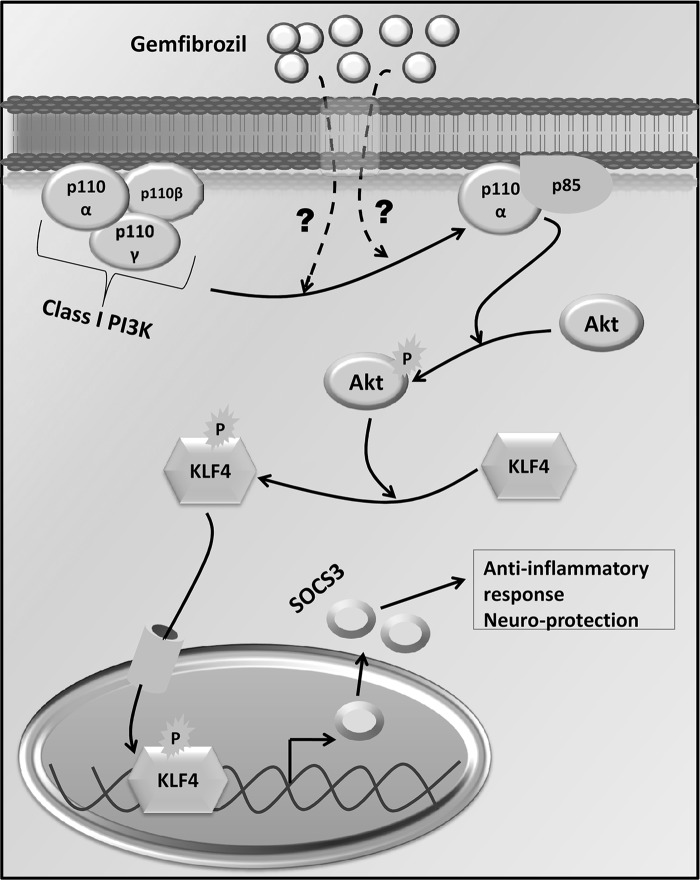
Glial inflammation is an important feature of several neurodegenerative disorders. Suppressor of cytokine signaling (SOCS) proteins play a crucial role in inhibiting cytokine signaling and inflammatory gene expression in various cell types, including glial cells. However, mechanisms by which SOCS genes could be up-regulated are poorly understood. This study underlines the importance of gemfibrozil, a Food and Drug Administration-approved lipid-lowering drug, in up-regulating the expression of SOCS3 in glial cells. Gemfibrozil increased the expression of Socs3 mRNA and protein in mouse astroglia and microglia in both a time- and dose-dependent manner. Interestingly, gemfibrozil induced the activation of type IA phosphatidylinositol (PI) 3-kinase and AKT. Accordingly, inhibition of PI 3-kinase and AKT by chemical inhibitors abrogated gemfibrozil-mediated up-regulation of SOCS3. Furthermore, we demonstrated that gemfibrozil induced the activation of Krüppel-like factor 4 (KLF4) via the PI 3-kinase-AKT pathway and that siRNA knockdown of KLF4 abrogated gemfibrozil-mediated up-regulation of SOCS3. Gemfibrozil also induced the recruitment of KLF4 to the distal, but not proximal, KLF4-binding site of the Socs3 promoter. This study delineates a novel property of gemfibrozil in up-regulating SOCS3 in glial cells via PI 3-kinase-AKT-mediated activation of KLF4 and suggests that gemfibrozil may find therapeutic application in neuroinflammatory and neurodegenerative disorders.
Figures



Similar articles
-
Gemfibrozil, a lipid-lowering drug, upregulates IL-1 receptor antagonist in mouse cortical neurons: implications for neuronal self-defense.J Immunol. 2012 Jul 15;189(2):1002-13. doi: 10.4049/jimmunol.1102624. Epub 2012 Jun 15. J Immunol. 2012. PMID: 22706077 Free PMC article.
-
Upregulation of Suppressor of Cytokine Signaling 3 in Microglia by Cinnamic Acid.Curr Alzheimer Res. 2018;15(10):894-904. doi: 10.2174/1567205015666180507104755. Curr Alzheimer Res. 2018. PMID: 29732971 Free PMC article.
-
Gemfibrozil, food and drug administration-approved lipid-lowering drug, increases longevity in mouse model of late infantile neuronal ceroid lipofuscinosis.J Neurochem. 2017 May;141(3):423-435. doi: 10.1111/jnc.13987. Epub 2017 Apr 3. J Neurochem. 2017. PMID: 28199020 Free PMC article.
-
[Research advances on relationship between suppressor of cytokine signaling 3 and myeloproliferative neoplasms].Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2010 Aug;18(4):1101-4. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2010. PMID: 20723338 Review. Chinese.
-
Gemfibrozil, stretching arms beyond lipid lowering.Immunopharmacol Immunotoxicol. 2009;31(3):339-51. doi: 10.1080/08923970902785253. Immunopharmacol Immunotoxicol. 2009. PMID: 19694602 Free PMC article. Review.
Cited by
-
Sodium phenylbutyrate enhances astrocytic neurotrophin synthesis via protein kinase C (PKC)-mediated activation of cAMP-response element-binding protein (CREB): implications for Alzheimer disease therapy.J Biol Chem. 2013 Mar 22;288(12):8299-8312. doi: 10.1074/jbc.M112.426536. Epub 2013 Feb 12. J Biol Chem. 2013. PMID: 23404502 Free PMC article.
-
Upregulation of IL-1 Receptor Antagonist by Aspirin in Glial Cells via Peroxisome Proliferator-Activated Receptor-Alpha.J Alzheimers Dis Rep. 2021 Aug 10;5(1):647-661. doi: 10.3233/ADR-210026. eCollection 2021. J Alzheimers Dis Rep. 2021. PMID: 34632302 Free PMC article.
-
Sodium Benzoate, a Metabolite of Cinnamon and a Food Additive, Upregulates Ciliary Neurotrophic Factor in Astrocytes and Oligodendrocytes.Neurochem Res. 2015 Nov;40(11):2333-47. doi: 10.1007/s11064-015-1723-x. Epub 2015 Sep 23. Neurochem Res. 2015. PMID: 26399250 Free PMC article.
-
Cinnamic acid, a natural plant compound, exhibits neuroprotection in a mouse model of Sandhoff disease via PPARα.NeuroImmune Pharm Ther. 2024 Mar 15;3(1):17-32. doi: 10.1515/nipt-2023-0027. eCollection 2024 Mar. NeuroImmune Pharm Ther. 2024. PMID: 38532783 Free PMC article.
-
PPARα in lysosomal biogenesis: A perspective.Pharmacol Res. 2016 Jan;103:144-8. doi: 10.1016/j.phrs.2015.11.011. Epub 2015 Nov 24. Pharmacol Res. 2016. PMID: 26621249 Free PMC article.
References
-
- Martin J. B. (1999) Molecular basis of the neurodegenerative disorders. N. Engl. J. Med. 340, 1970–1980 - PubMed
-
- Cummings J. L., Vinters H. V., Cole G. M., Khachaturian Z. S. (1998) Alzheimer disease. Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 51, S2–S17 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
