

Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and δ in brown adipocytes to match fatty acid oxidation with supply
- PMID: 22685301
- PMCID: PMC3408177
- DOI: 10.1074/jbc.M112.374041
Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and δ in brown adipocytes to match fatty acid oxidation with supply
Abstract
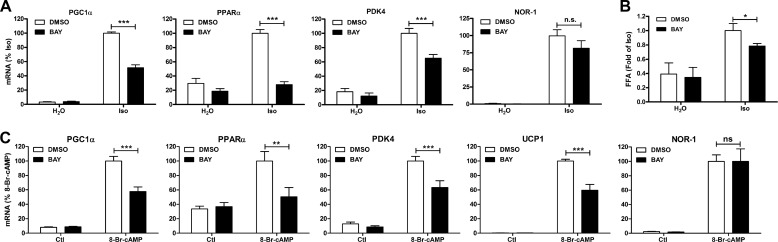
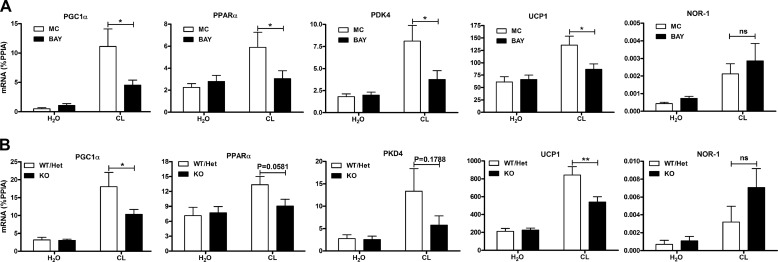
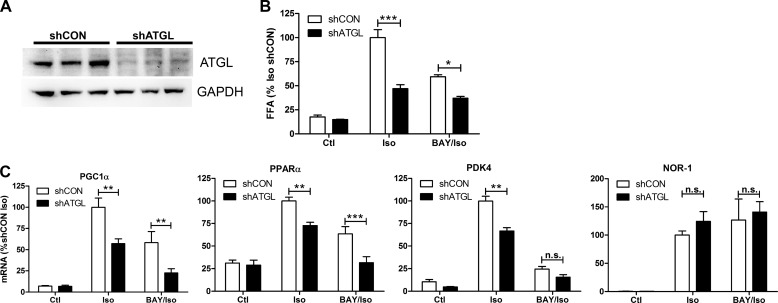
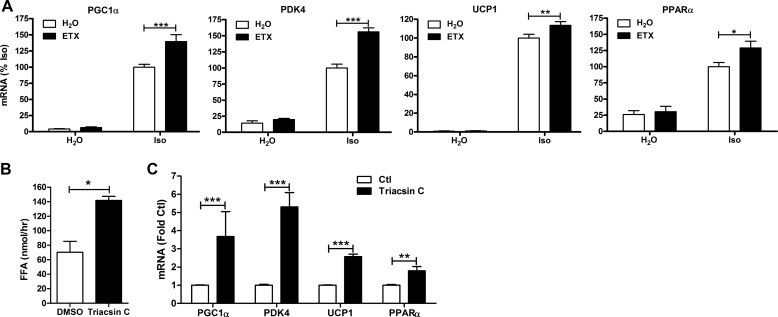
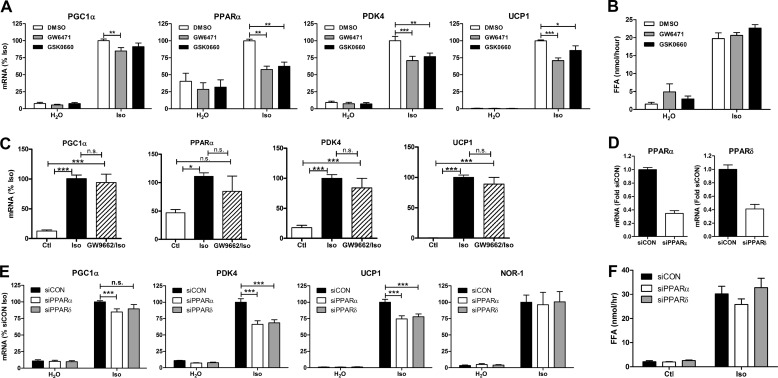
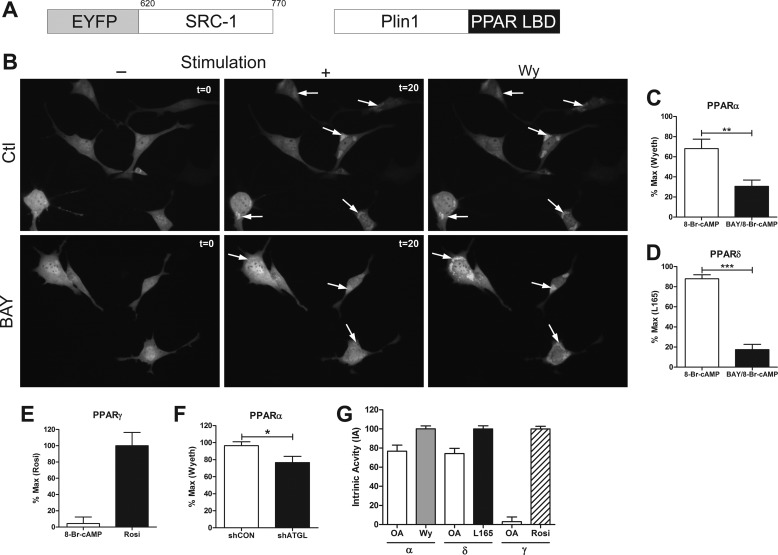
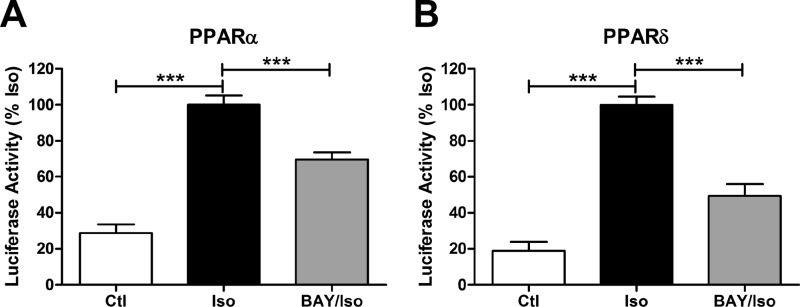
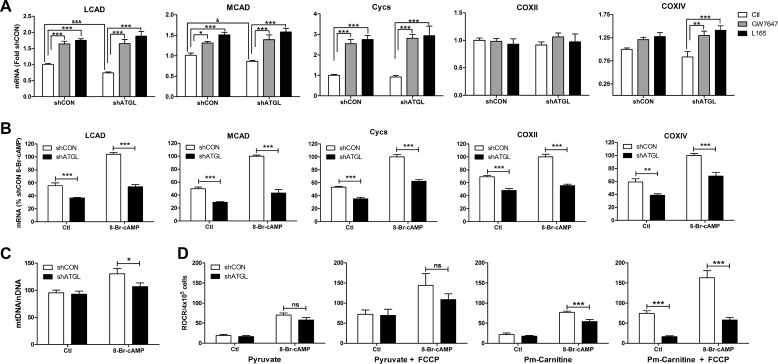
β-Adrenergic receptors (β-ARs) promote brown adipose tissue (BAT) thermogenesis by mobilizing fatty acids and inducing the expression of oxidative genes. β-AR activation increases the expression of oxidative genes by elevating cAMP, but whether lipolytic products can modulate gene expression is not known. This study examined the role that adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) plays in the induction of gene expression. Activation of brown adipocytes by β-AR agonism or 8-bromo-cyclic AMP increased the expression of PGC1α, PDK4, PPARα, uncoupling protein 1 (UCP1), and neuron-derived orphan receptor-1 (NOR-1), and concurrent inhibition of HSL reduced the induction of PGC1α, PDK4, PPARα, and UCP1 but not NOR-1. Similar results were observed in the BAT of mice following pharmacological or genetic inhibition of HSL and in brown adipocytes with stable knockdown of ATGL. Conversely, treatments that increase endogenous fatty acids elevated the expression of oxidative genes. Pharmacological antagonism and siRNA knockdown indicate that PPARα and PPARδ modulate the induction of oxidative genes by β-AR agonism. Using a live cell fluorescent reporter assay of PPAR activation, we demonstrated that ligands for PPARα and -δ, but not PPARγ, were rapidly generated at the lipid droplet surface and could transcriptionally activate PPARα and -δ. Knockdown of ATGL reduced cAMP-mediated induction of genes involved in fatty acid oxidation and oxidative phosphorylation. Consequently, ATGL knockdown reduced maximal oxidation of fatty acids, but not pyruvate, in response to cAMP stimulation. Overall, the results indicate that lipolytic products can activate PPARα and PPARδ in brown adipocytes, thereby expanding the oxidative capacity to match enhanced fatty acid supply.
Figures
References
-
- Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 - PubMed
-
- Spiegelman B. M. (2007) Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found. Symp. 287, 60–63; discussion 63–69 - PubMed
-
- Rim J. S., Kozak L. P. (2002) Regulatory motifs for CREB-binding protein and Nfe2l2 transcription factors in the upstream enhancer of the mitochondrial uncoupling protein 1 gene. J. Biol. Chem. 277, 34589–34600 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
