

Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma
- PMID: 22685613
- PMCID: PMC3369837
- DOI: 10.1371/journal.pone.0038863
Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma
Abstract
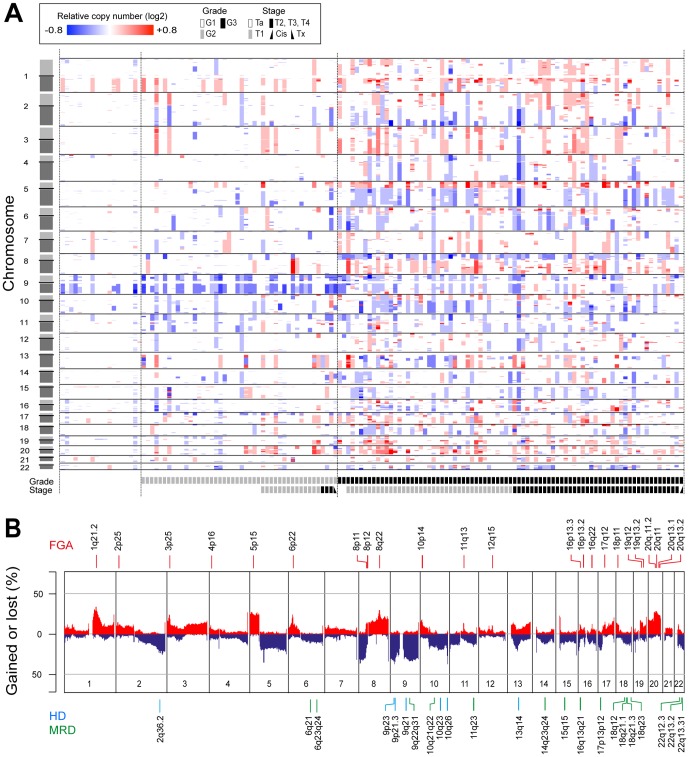
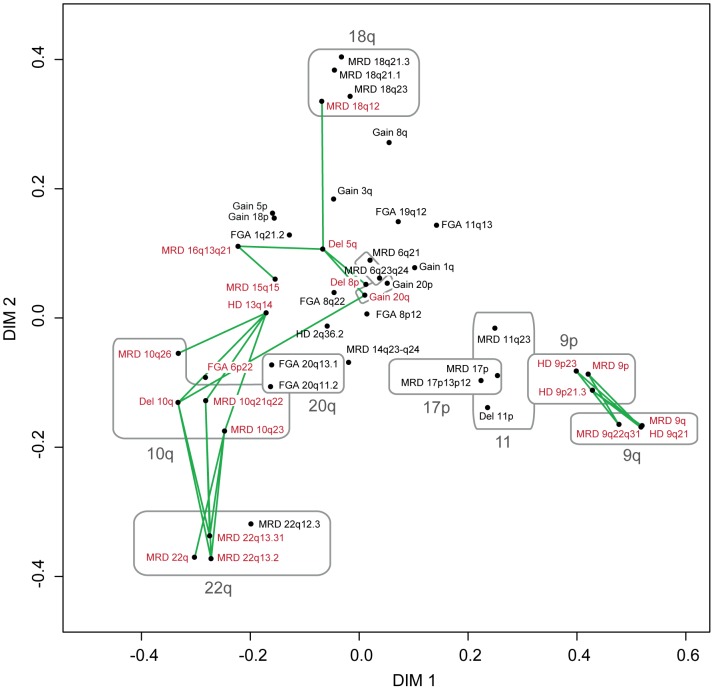
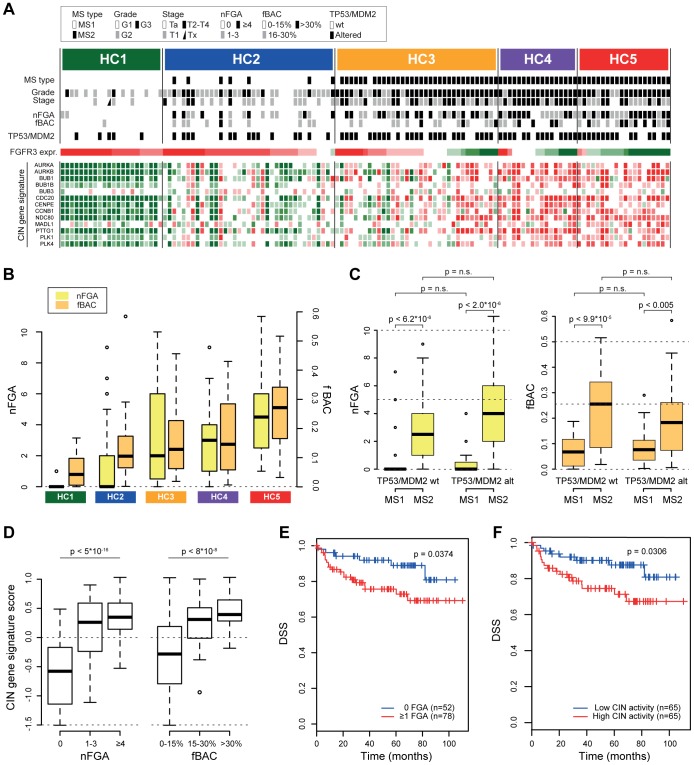
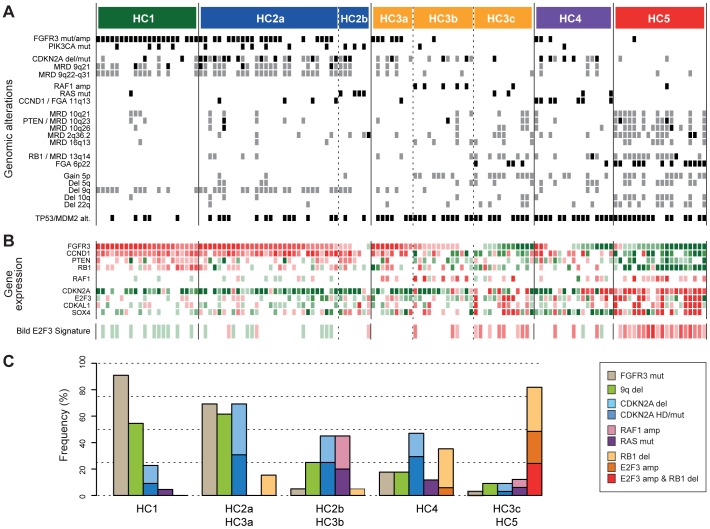
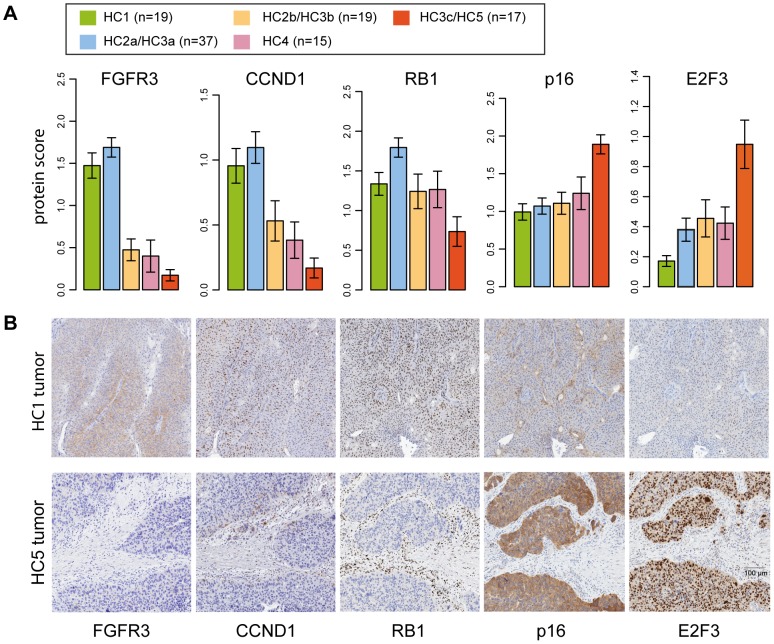
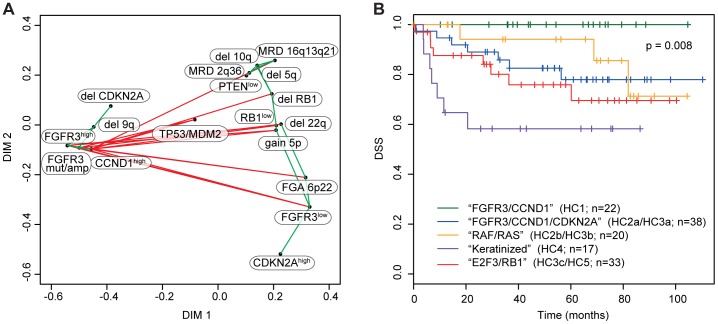
Similar to other malignancies, urothelial carcinoma (UC) is characterized by specific recurrent chromosomal aberrations and gene mutations. However, the interconnection between specific genomic alterations, and how patterns of chromosomal alterations adhere to different molecular subgroups of UC, is less clear. We applied tiling resolution array CGH to 146 cases of UC and identified a number of regions harboring recurrent focal genomic amplifications and deletions. Several potential oncogenes were included in the amplified regions, including known oncogenes like E2F3, CCND1, and CCNE1, as well as new candidate genes, such as SETDB1 (1q21), and BCL2L1 (20q11). We next combined genome profiling with global gene expression, gene mutation, and protein expression data and identified two major genomic circuits operating in urothelial carcinoma. The first circuit was characterized by FGFR3 alterations, overexpression of CCND1, and 9q and CDKN2A deletions. The second circuit was defined by E3F3 amplifications and RB1 deletions, as well as gains of 5p, deletions at PTEN and 2q36, 16q, 20q, and elevated CDKN2A levels. TP53/MDM2 alterations were common for advanced tumors within the two circuits. Our data also suggest a possible RAS/RAF circuit. The tumors with worst prognosis showed a gene expression profile that indicated a keratinized phenotype. Taken together, our integrative approach revealed at least two separate networks of genomic alterations linked to the molecular diversity seen in UC, and that these circuits may reflect distinct pathways of tumor development.
Conflict of interest statement
Figures
References
-
- Richter J, Jiang F, Gorog JP, Sartorius G, Egenter C. Marked genetic differences between stage pTa and stage pT1 papillary bladder cancer detected by comparative genomic hybridization. Cancer research. 1997;57:2864. - PubMed
-
- Hoglund M, Sall T, Heim S, Mitelman F, Mandahl N. Identification of cytogenetic subgroups and karyotypic pathways in transitional cell carcinoma. Cancer research. 2001;61:8246. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
