

SEQuel: improving the accuracy of genome assemblies
- PMID: 22689760
- PMCID: PMC3371851
- DOI: 10.1093/bioinformatics/bts219
SEQuel: improving the accuracy of genome assemblies
Abstract
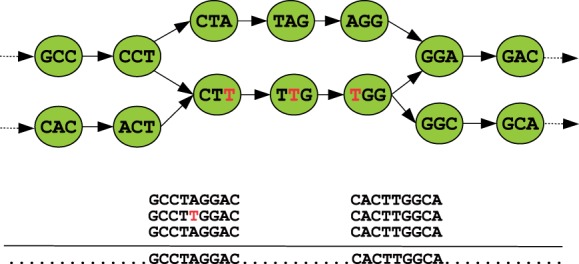
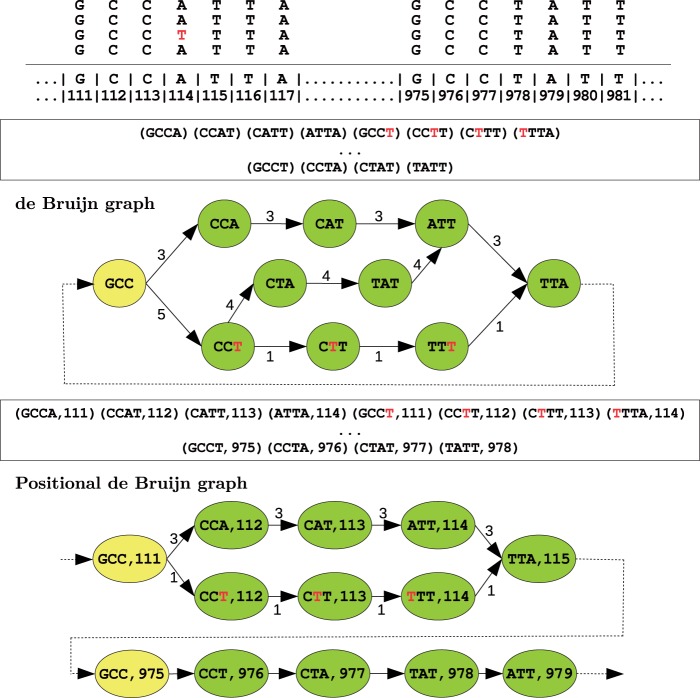
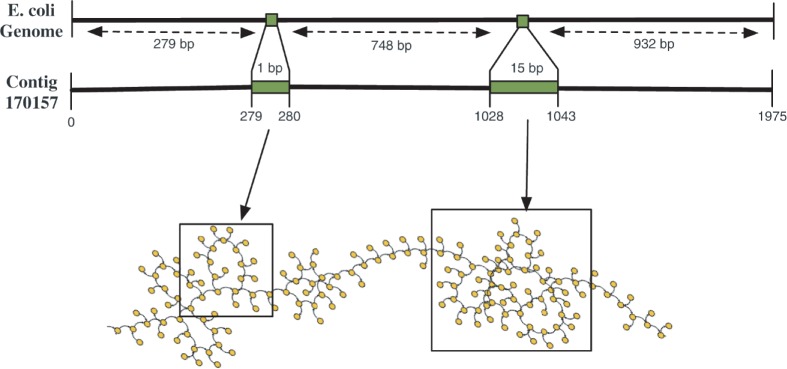
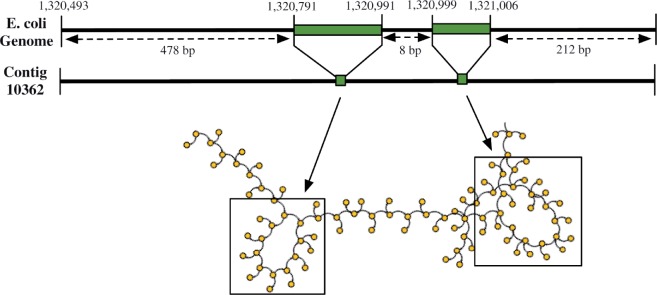
Motivation: Assemblies of next-generation sequencing (NGS) data, although accurate, still contain a substantial number of errors that need to be corrected after the assembly process. We develop SEQuel, a tool that corrects errors (i.e. insertions, deletions and substitution errors) in the assembled contigs. Fundamental to the algorithm behind SEQuel is the positional de Bruijn graph, a graph structure that models k-mers within reads while incorporating the approximate positions of reads into the model.
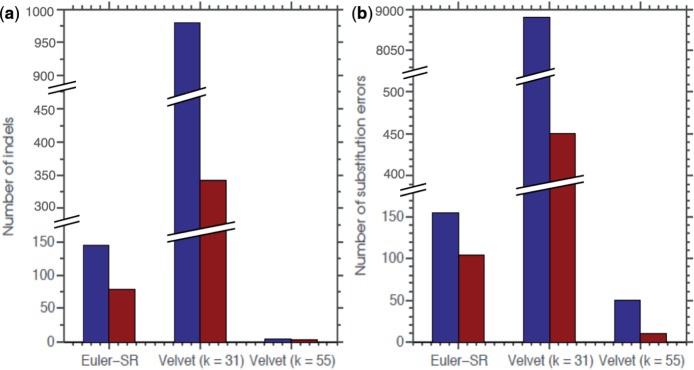
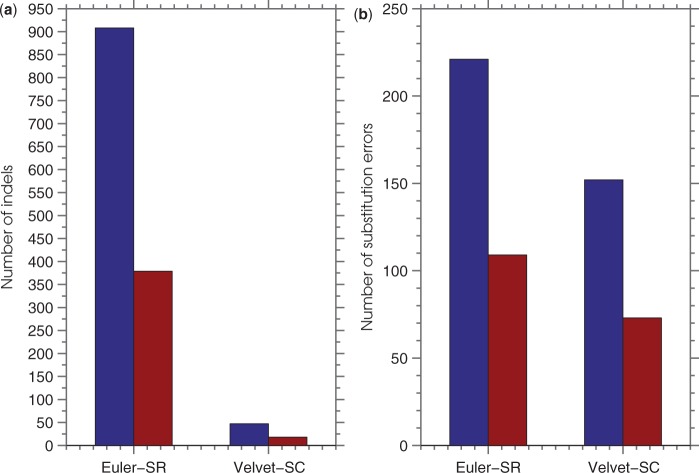
Results: SEQuel reduced the number of small insertions and deletions in the assemblies of standard multi-cell Escherichia coli data by almost half, and corrected between 30% and 94% of the substitution errors. Further, we show SEQuel is imperative to improving single-cell assembly, which is inherently more challenging due to higher error rates and non-uniform coverage; over half of the small indels, and substitution errors in the single-cell assemblies were corrected. We apply SEQuel to the recently assembled Deltaproteobacterium SAR324 genome, which is the first bacterial genome with a comprehensive single-cell genome assembly, and make over 800 changes (insertions, deletions and substitutions) to refine this assembly.
Availability: SEQuel can be used as a post-processing step in combination with any NGS assembler and is freely available at http://bix.ucsd.edu/SEQuel/.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
