

Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice
- PMID: 22689975
- PMCID: PMC3387061
- DOI: 10.1073/pnas.1202071109
Arylsulfatase G inactivation causes loss of heparan sulfate 3-O-sulfatase activity and mucopolysaccharidosis in mice
Abstract
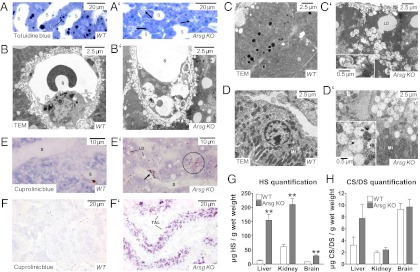
Deficiency of glycosaminoglycan (GAG) degradation causes a subclass of lysosomal storage disorders called mucopolysaccharidoses (MPSs), many of which present with severe neuropathology. Critical steps in the degradation of the GAG heparan sulfate remain enigmatic. Here we show that the lysosomal arylsulfatase G (ARSG) is the long-sought glucosamine-3-O-sulfatase required to complete the degradation of heparan sulfate. Arsg-deficient mice accumulate heparan sulfate in visceral organs and the central nervous system and develop neuronal cell death and behavioral deficits. This accumulated heparan sulfate exhibits unique nonreducing end structures with terminal N-sulfoglucosamine-3-O-sulfate residues, allowing diagnosis of the disorder. Recombinant human ARSG is able to cleave 3-O-sulfate groups from these residues as well as from an authentic 3-O-sulfated N-sulfoglucosamine standard. Our results demonstrate the key role of ARSG in heparan sulfate degradation and strongly suggest that ARSG deficiency represents a unique, as yet unknown form of MPS, which we term MPS IIIE.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Ballabio A, Gieselmann V. Lysosomal disorders: From storage to cellular damage. Biochim Biophys Acta. 2009;1793:684–696. - PubMed
-
- Diez-Roux G, Ballabio A. Sulfatases and human disease. Annu Rev Genomics Hum Genet. 2005;6:355–379. - PubMed
-
- Mehl E, Jatzkewitz H. Cerebroside 3-sulfate as a physiological substrate of arylsulfatase A. Biochim Biophys Acta. 1968;151:619–627. - PubMed
-
- von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: Scriver CR, et al., editors. The Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill; 2001. pp. 3694–3724.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
