

Differences in local genomic context of bound and unbound motifs
- PMID: 22692006
- PMCID: PMC3412921
- DOI: 10.1016/j.gene.2012.06.005
Differences in local genomic context of bound and unbound motifs
Abstract
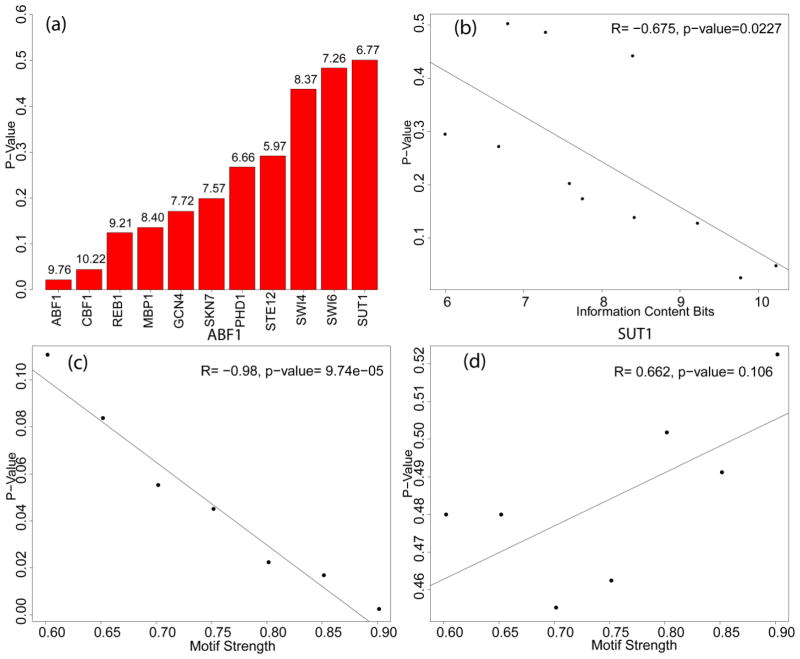
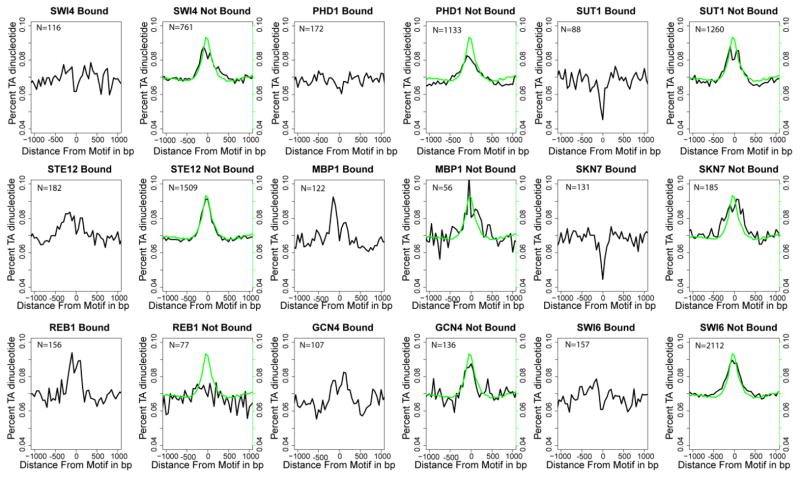
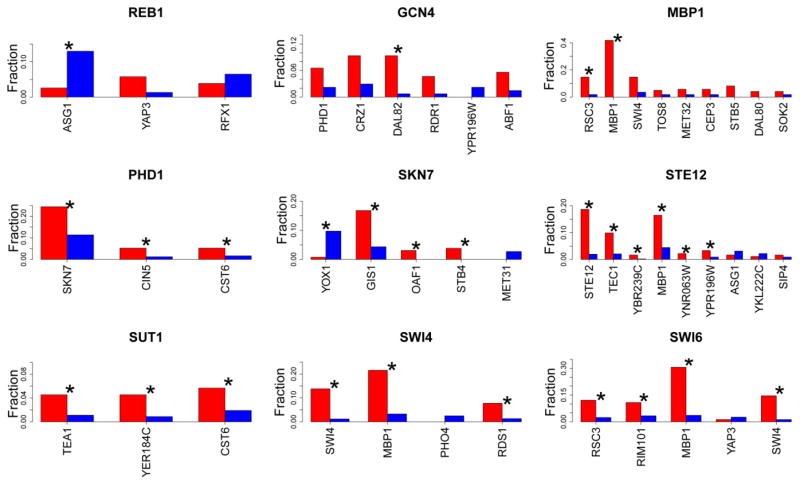
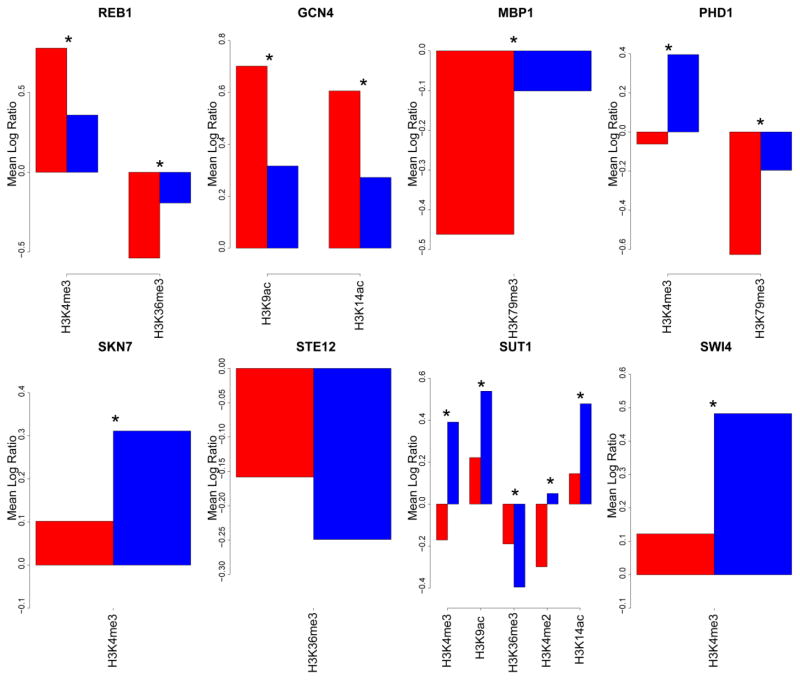
Understanding gene regulation is a major objective in molecular biology research. Frequently, transcription is driven by transcription factors (TFs) that bind to specific DNA sequences. These motifs are usually short and degenerate, rendering the likelihood of multiple copies occurring throughout the genome due to random chance as high. Despite this, TFs only bind to a small subset of sites, thus prompting our investigation into the differences between motifs that are bound by TFs and those that remain unbound. Here we constructed vectors representing various chromatin- and sequence-based features for a published set of bound and unbound motifs representing nine TFs in the budding yeast Saccharomyces cerevisiae. Using a machine learning approach, we identified a set of features that can be used to discriminate between bound and unbound motifs. We also discovered that some TFs bind most or all of their strong motifs in intergenic regions. Our data demonstrate that local sequence context can be strikingly different around motifs that are bound compared to motifs that are unbound. We concluded that there are multiple combinations of genomic features that characterize bound or unbound motifs.
Published by Elsevier B.V.
Figures
References
-
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, Gebbia M, Talukder S, Yang A, Mnaimneh S, Terterov D, Coburn D, Li Yeo A, Yeo ZX, Clarke ND, Lieb JD, Ansari AZ, Nislow C, Hughes TR. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Molecular Cell. 2008;32:878–87. - PMC - PubMed
-
- Benjamini Y, Yekutieli D. The Control of the False Discovery Rate in Multiple Testing under Dependency. The Annals of Statistics. 2001;29:1165–1188.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
