

Review
doi: 10.1038/ejhg.2012.100.
Epub 2012 Jun 13.
Aniridia
Affiliations
- PMID: 22692063
- PMCID: PMC3449076
- DOI: 10.1038/ejhg.2012.100
Item in Clipboard
Review
Aniridia
Eur J Hum Genet.
2012 Oct.
Abstract
Aniridia is a rare congenital disorder in which there is a variable degree of hypoplasia or the absence of iris tissue associated with multiple other ocular changes, some present from birth and some arising progressively over time. Most cases are associated with dominantly inherited mutations or deletions of the PAX6 gene. This article will review the clinical manifestations, the molecular basis including genotype-phenotype correlations, diagnostic approaches and management of aniridia.
Figures
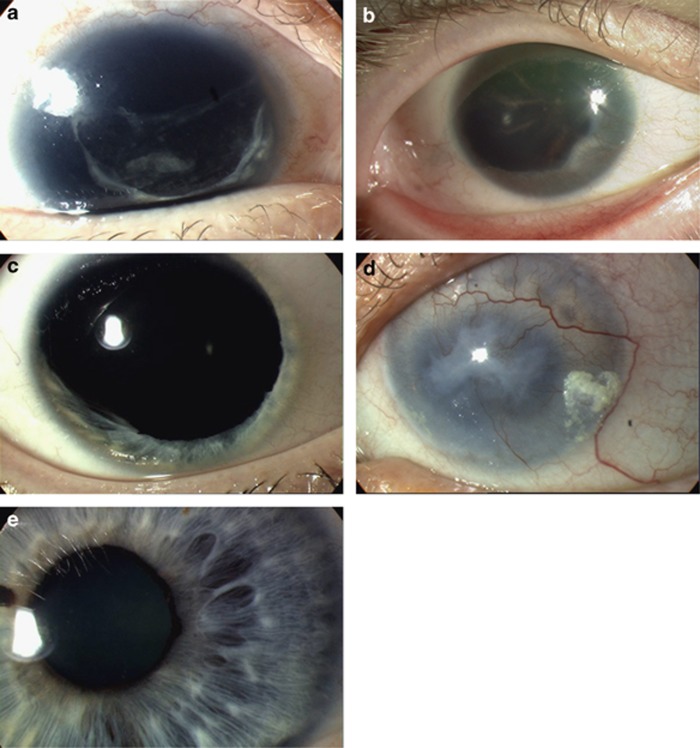
Heterozygous PAX6 mutations cause a range of eye phenotypes. (a) Total absence of iris tissue with surgical aphakia (removal of natural lens) and remnant of opaque lens capsule (CTE mutation). (b) Almost complete absence of iris tissue obscured by peripheral corneal changes with opacification and neovasularisation, and unusual partly pigmented cortical lens opacities (PTC mutation). (c) Partial absence of iris tissue (missense mutation). (d) Severe aniridic keratopathy with total loss of transparency and neovasularisation of the cornea (PTC mutation). (e) Full iris demonstrating substantial area of abnormal iris architecture (missense mutation). Panels (c) and (e) are reproduced from Hingorani et al. Association for Research in Vision and Ophthalmology.
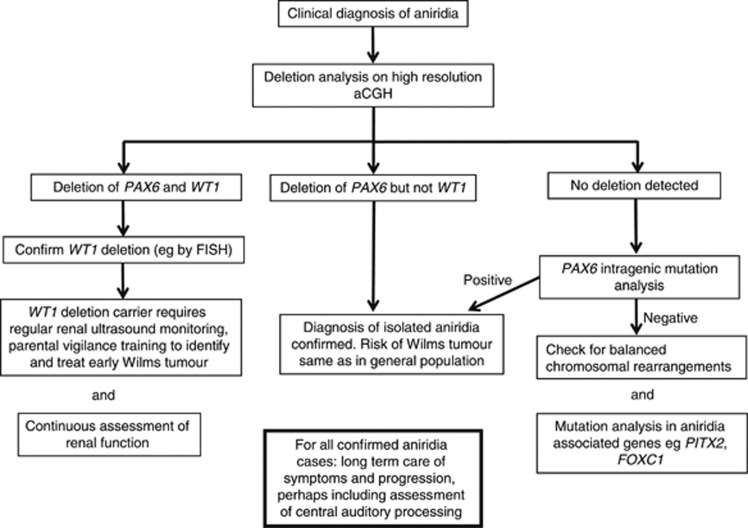
Flow-chart illustrating a suggested strategy for molecular analysis of aniridia cases. The priority is to determine whether there is an underlying WAGR deletion (involving both the PAX6 and WT1 loci), which strongly predisposes to Wilms tumour. In cases where WT1 is not deleted, detection of a PAX6 mutation is less urgent but provides a sound basis for genetic counselling. This diagram is intended to show general principles only – molecular techniques are evolving quickly and the specific approaches mentioned here may be superseded by more efficient and cost-effective methods.
References
-
- Nelson LB, Spaeth GL, Nowinski TS, Margo CE, Jackson L. Aniridia. A review. Surv Ophthalmol. 1984;28:621–642. - PubMed
-
- Lee H, Khan R, O'Keefe M. Aniridia: current pathology and management. Acta Ophthalmol. 2008;86:708–715. - PubMed
-
- Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics. 2005;116:984–988. - PubMed
-
- Gronskov K, Olsen JH, Sand A, et al. Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet. 2001;109:11–18. - PubMed
-
- Valenzuela A, Cline RA. Ocular and nonocular findings in patients with aniridia. Can J Ophthalmol. 2004;39:632–638. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
