

Activation of transient receptor potential canonical 3 (TRPC3)-mediated Ca2+ entry by A1 adenosine receptor in cardiomyocytes disturbs atrioventricular conduction
- PMID: 22692208
- PMCID: PMC3411008
- DOI: 10.1074/jbc.M112.378588
Activation of transient receptor potential canonical 3 (TRPC3)-mediated Ca2+ entry by A1 adenosine receptor in cardiomyocytes disturbs atrioventricular conduction
Abstract
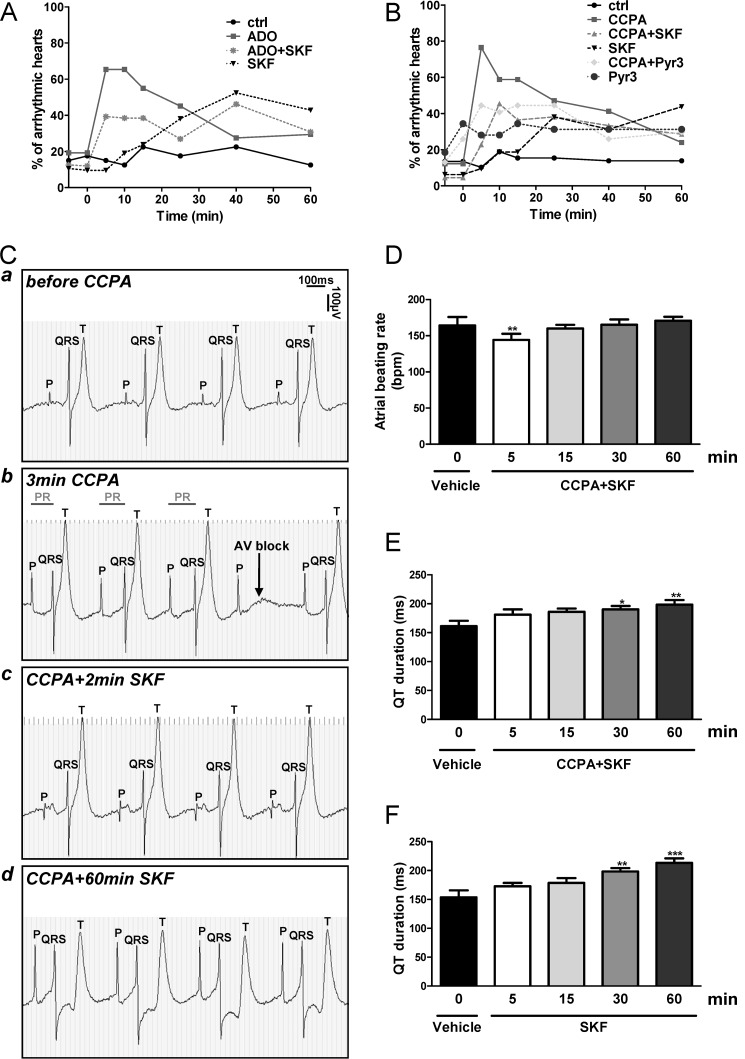
Although the activation of the A(1)-subtype of the adenosine receptors (A(1)AR) is arrhythmogenic in the developing heart, little is known about the underlying downstream mechanisms. The aim of this study was to determine to what extent the transient receptor potential canonical (TRPC) channel 3, functioning as receptor-operated channel (ROC), contributes to the A(1)AR-induced conduction disturbances. Using embryonic atrial and ventricular myocytes obtained from 4-day-old chick embryos, we found that the specific activation of A(1)AR by CCPA induced sarcolemmal Ca(2+) entry. However, A(1)AR stimulation did not induce Ca(2+) release from the sarcoplasmic reticulum. Specific blockade of TRPC3 activity by Pyr3, by a dominant negative of TRPC3 construct, or inhibition of phospholipase Cs and PKCs strongly inhibited the A(1)AR-enhanced Ca(2+) entry. Ca(2+) entry through TRPC3 was activated by the 1,2-diacylglycerol (DAG) analog OAG via PKC-independent and -dependent mechanisms in atrial and ventricular myocytes, respectively. In parallel, inhibition of the atypical PKCζ by myristoylated PKCζ pseudosubstrate inhibitor significantly decreased the A(1)AR-enhanced Ca(2+) entry in both types of myocytes. Additionally, electrocardiography showed that inhibition of TRPC3 channel suppressed transient A(1)AR-induced conduction disturbances in the embryonic heart. Our data showing that A(1)AR activation subtly mediates a proarrhythmic Ca(2+) entry through TRPC3-encoded ROC by stimulating the phospholipase C/DAG/PKC cascade provide evidence for a novel pathway whereby Ca(2+) entry and cardiac function are altered. Thus, the A(1)AR-TRPC3 axis may represent a potential therapeutic target.
Figures







References
-
- Görlach A. (2005) Control of adenosine transport by hypoxia. Circ. Res. 97, 1–3 - PubMed
-
- Peart J., Headrick J. P. (2000) Intrinsic A1 adenosine receptor activation during ischemia or reperfusion improves recovery in mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 279, H2166–2175 - PubMed
-
- Roscoe A. K., Christensen J. D., Lynch C., 3rd (2000) Isoflurane, but not halothane, induces protection of human myocardium via adenosine A1 receptors and adenosine triphosphate-sensitive potassium channels. Anesthesiology 92, 1692–1701 - PubMed
-
- Cerniway R. J., Morrison R. R., Byford A. M., Lankford A. R., Headrick J. P., Van Wylen D. G., Matherne G. P. (2002) A1 adenosine receptor overexpression decreases stunning from anoxia-reoxygenation: role of the mitochondrial KATP channel. Basic Res. Cardiol. 97, 232–238 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
