

Bats worldwide carry hepatitis E virus-related viruses that form a putative novel genus within the family Hepeviridae
- PMID: 22696648
- PMCID: PMC3416139
- DOI: 10.1128/JVI.00800-12
Bats worldwide carry hepatitis E virus-related viruses that form a putative novel genus within the family Hepeviridae
Abstract
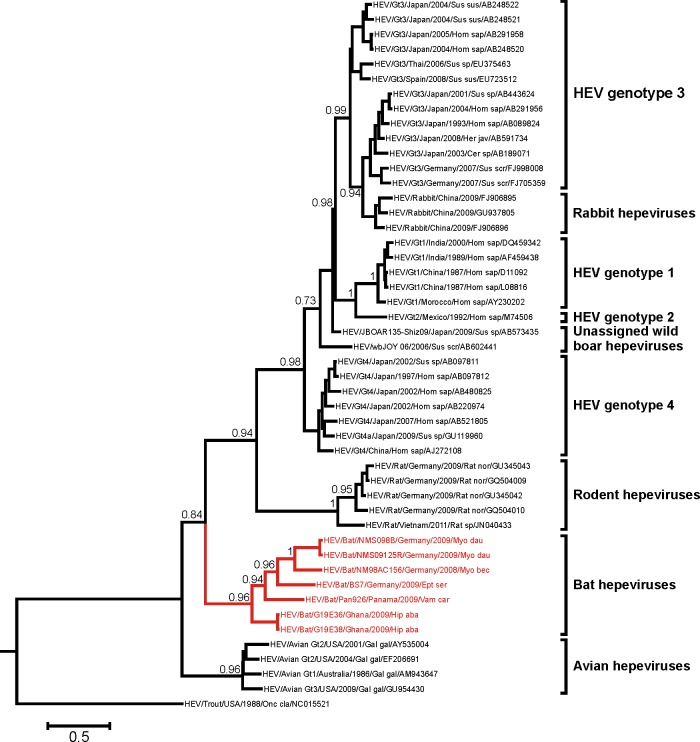
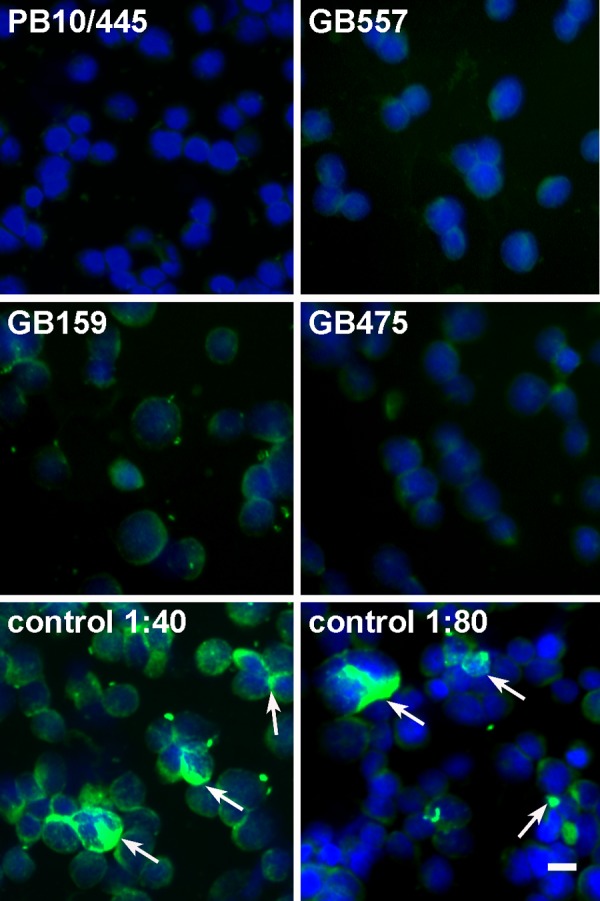
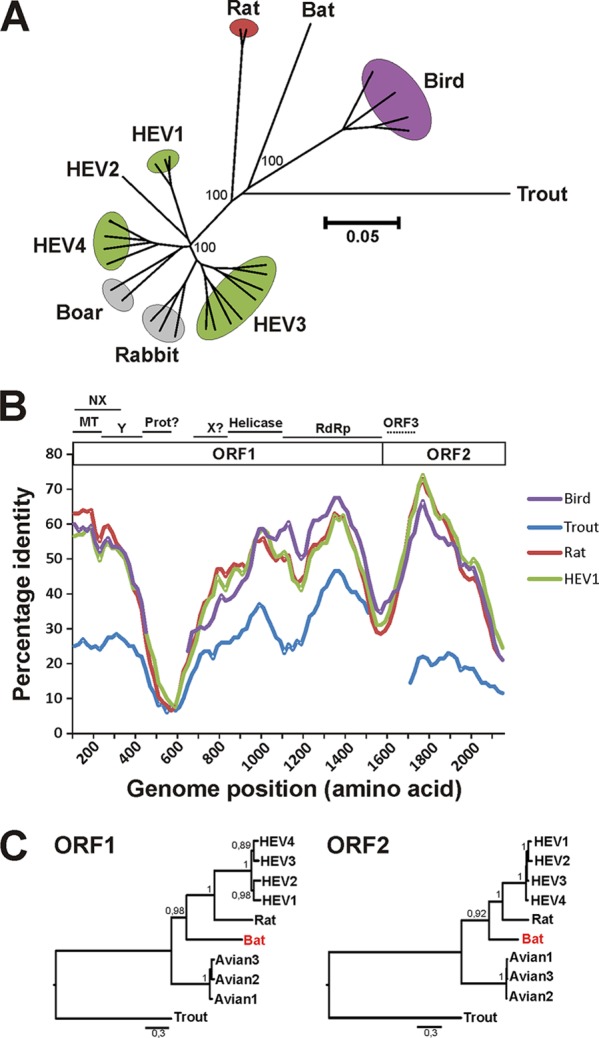
Hepatitis E virus (HEV) is one of the most common causes of acute hepatitis in tropical and temperate climates. Tropical genotypes 1 and 2 are associated with food-borne and waterborne transmission. Zoonotic reservoirs (mainly pigs, wild boar, and deer) are considered for genotypes 3 and 4, which exist in temperate climates. In view of the association of several zoonotic viruses with bats, we analyzed 3,869 bat specimens from 85 different species and from five continents for hepevirus RNA. HEVs were detected in African, Central American, and European bats, forming a novel phylogenetic clade in the family Hepeviridae. Bat hepeviruses were highly diversified and comparable to human HEV in sequence variation. No evidence for the transmission of bat hepeviruses to humans was found in over 90,000 human blood donations and individual patient sera. Full-genome analysis of one representative virus confirmed formal classification within the family Hepeviridae. Sequence- and distance-based taxonomic evaluations suggested that bat hepeviruses constitute a distinct genus within the family Hepeviridae and that at least three other genera comprising human, rodent, and avian hepeviruses can be designated. This may imply that hepeviruses invaded mammalian hosts nonrecently and underwent speciation according to their host restrictions. Human HEV-related viruses in farmed and peridomestic animals might represent secondary acquisitions of human viruses, rather than animal precursors causally involved in the evolution of human HEV.
Figures



References
-
- Adlhoch C, Kaiser M, Pauli G, Koch J, Meisel H. 2009. Indigenous hepatitis E virus infection of a plasma donor in Germany. Vox Sang. 97:303–308 - PubMed
-
- Batts W, Yun S, Hedrick R, Winton J. 2011. A novel member of the family Hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus Res. 158:116–123 - PubMed
-
- Baylis SA, Gartner T, Nick S, Ovemyr J, Blumel J. 2012. Occurrence of hepatitis E virus RNA in plasma donations from Sweden, Germany and the United States. Vox Sang. 103:89–90 - PubMed
-
- Baylis SA, Koc O, Nick S, Blumel J. 2012. Widespread distribution of hepatitis E virus in plasma fractionation pools. Vox Sang. 102:182–183 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
