

Brain-derived neurotrophic factor activation of CaM-kinase kinase via transient receptor potential canonical channels induces the translation and synaptic incorporation of GluA1-containing calcium-permeable AMPA receptors
- PMID: 22699894
- PMCID: PMC3390208
- DOI: 10.1523/JNEUROSCI.6034-11.2012
Brain-derived neurotrophic factor activation of CaM-kinase kinase via transient receptor potential canonical channels induces the translation and synaptic incorporation of GluA1-containing calcium-permeable AMPA receptors
Abstract
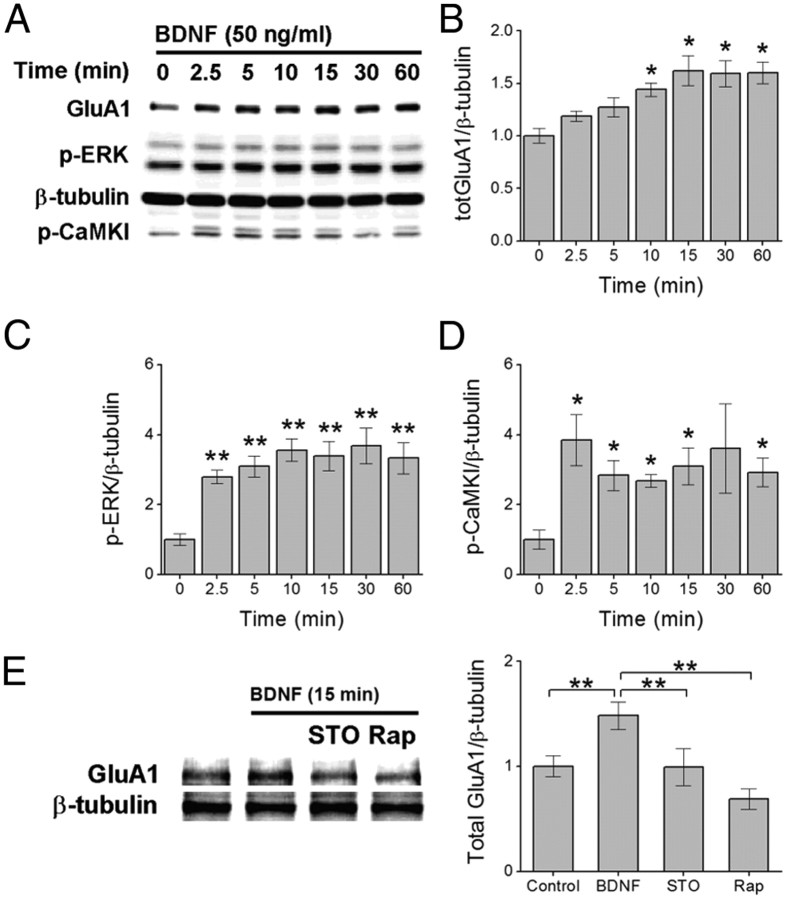
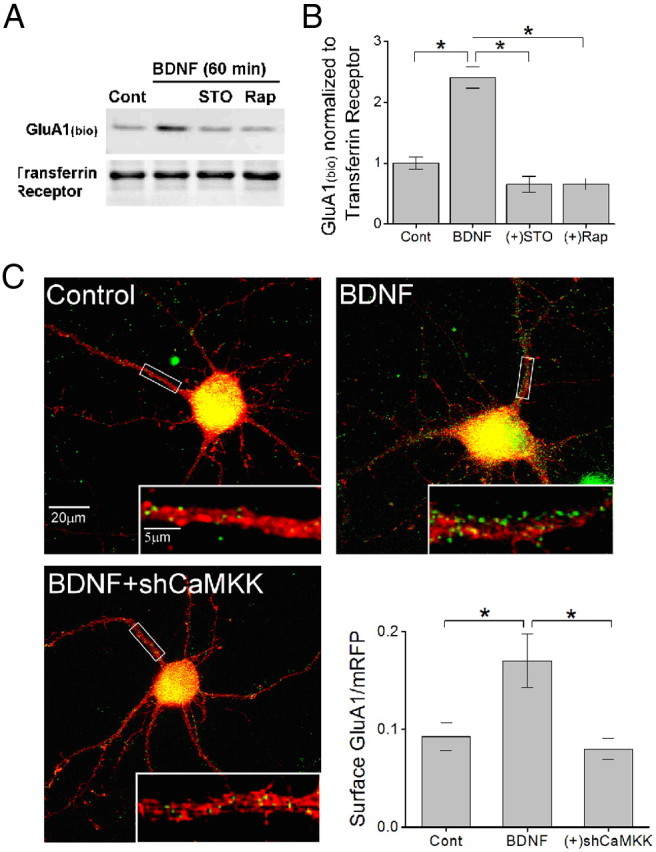
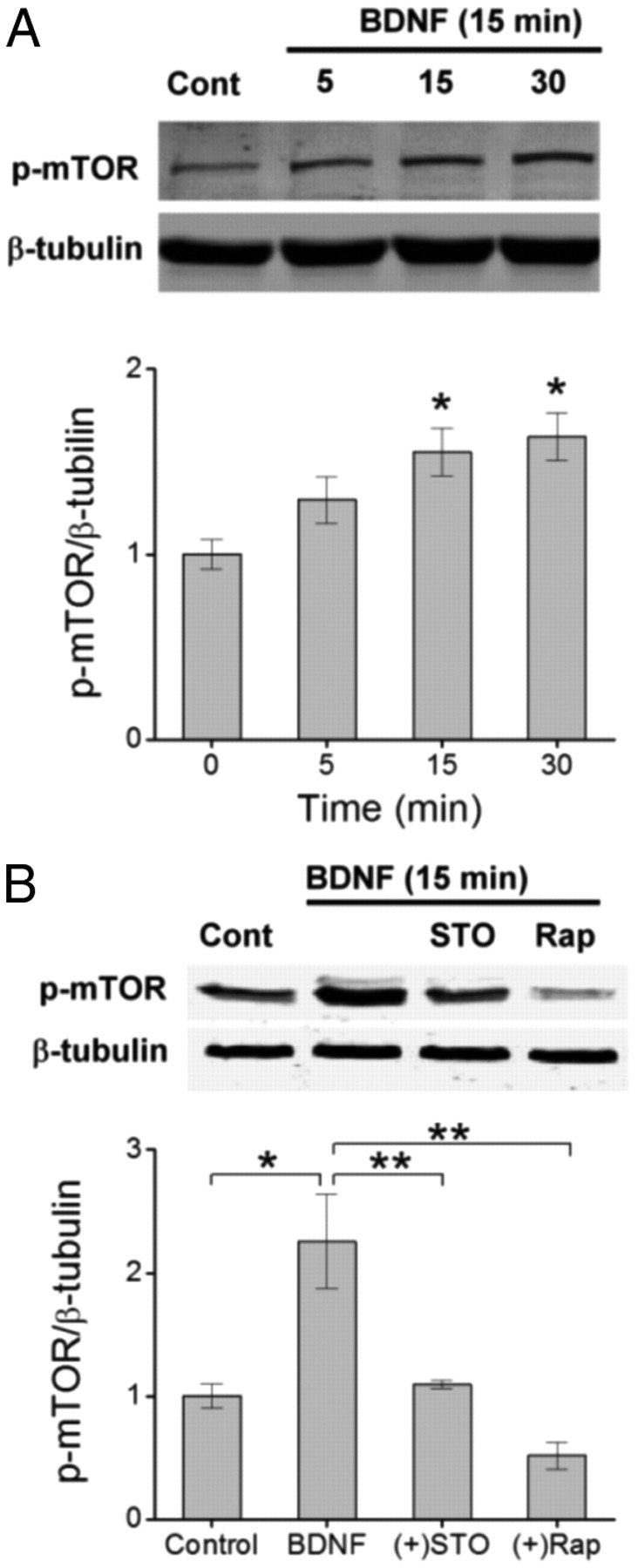
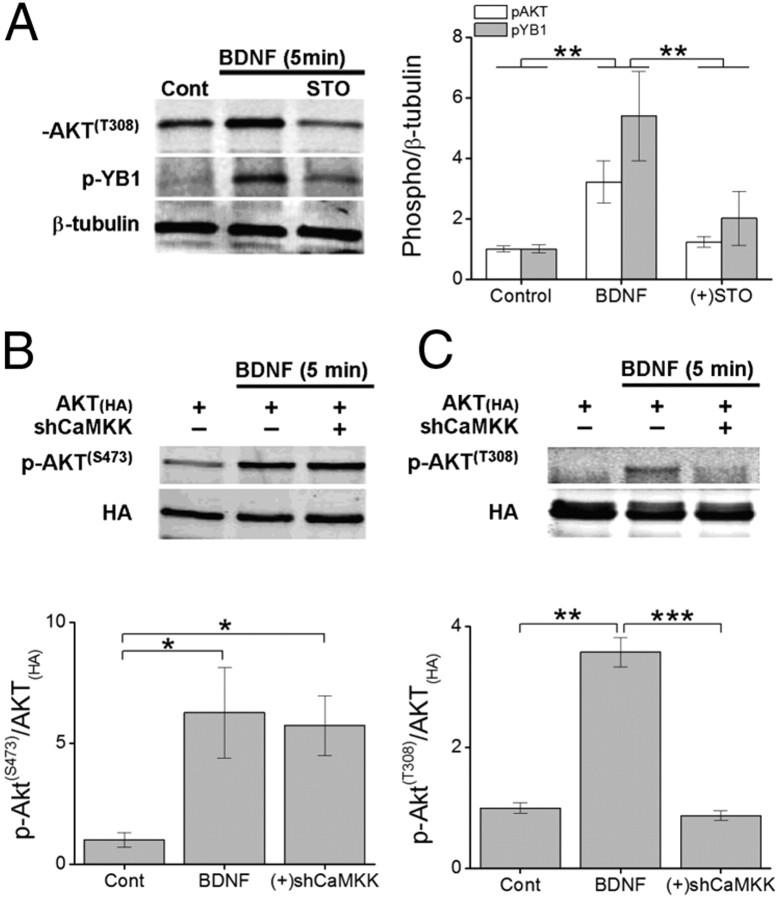
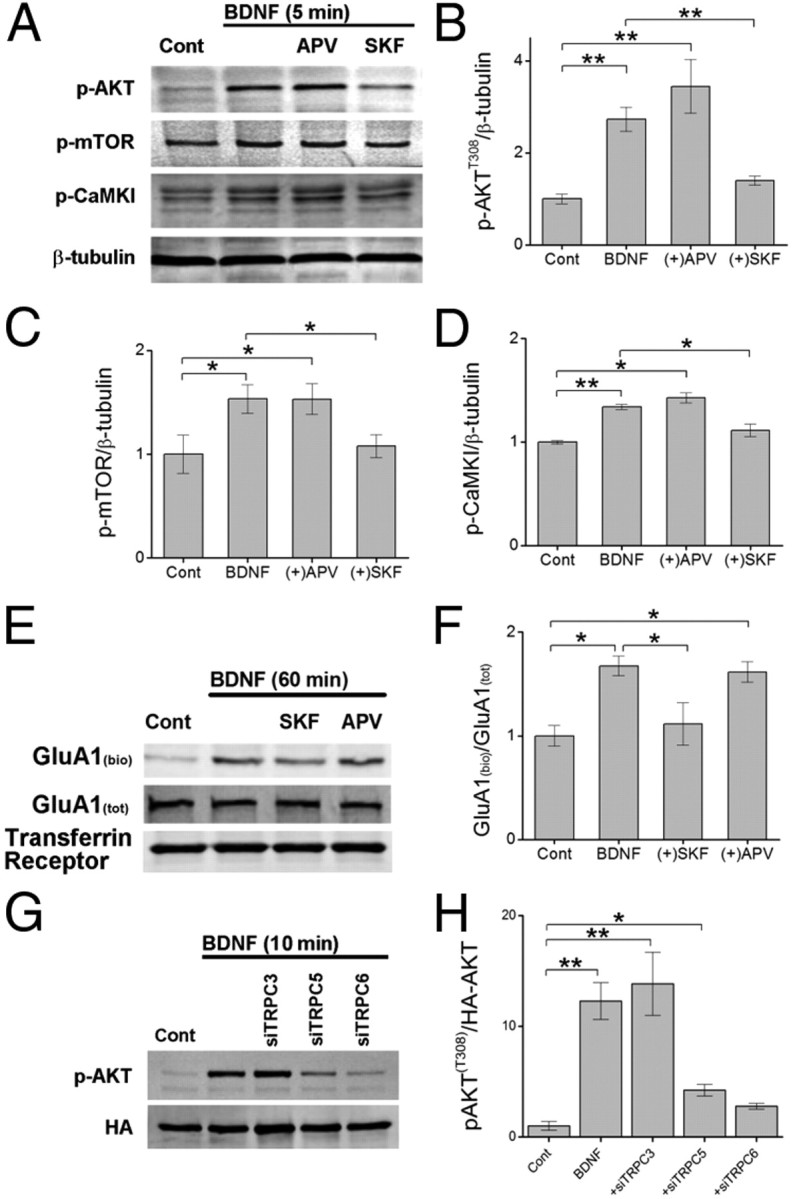
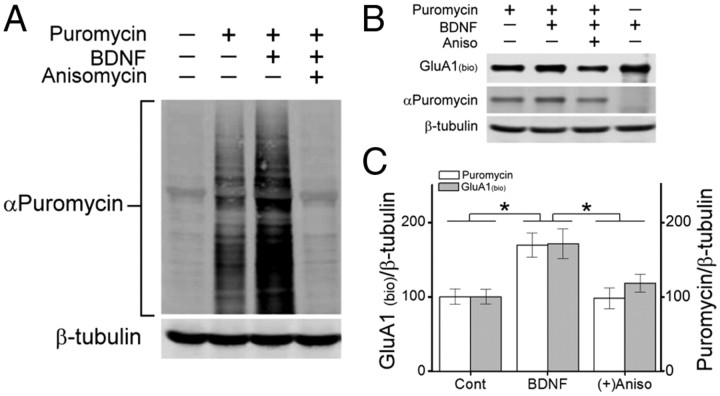
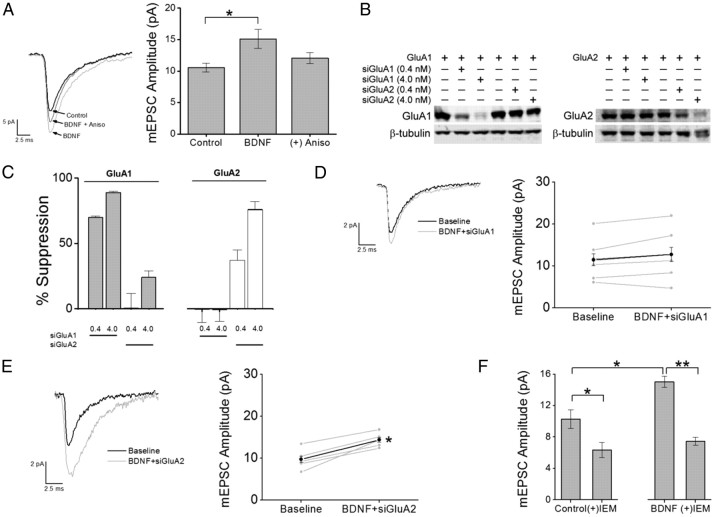
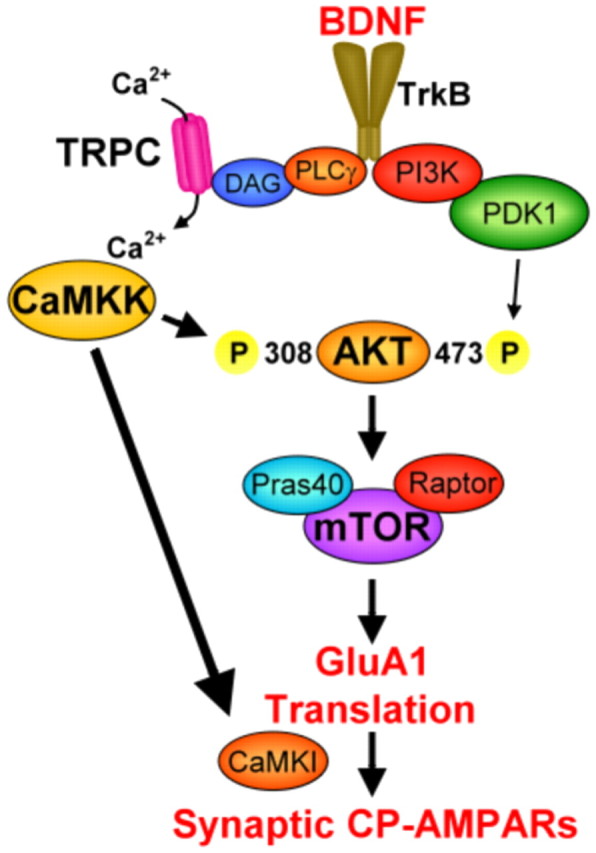
Glutamatergic synapses in early postnatal development transiently express calcium-permeable AMPA receptors (CP-AMPARs). Although these GluA2-lacking receptors are essential and are elevated in response to brain-derived neurotrophic factor (BDNF), little is known regarding molecular mechanisms that govern their expression and synaptic insertion. Here we show that BDNF-induced GluA1 translation in rat primary hippocampal neurons requires the activation of mammalian target of rapamycin (mTOR) via calcium calmodulin-dependent protein kinase kinase (CaMKK). Specifically, BDNF-mediated phosphorylation of threonine 308 (T308) in AKT, a known substrate of CaMKK and an upstream activator of mTOR-dependent translation, was prevented by (1) pharmacological inhibition of CaMKK with STO-609, (2) overexpression of a dominant-negative CaMKK, or (3) short hairpin-mediated knockdown of CaMKK. GluA1 surface expression induced by BDNF, as assessed by immunocytochemistry using an extracellular N-terminal GluA1 antibody or by surface biotinylation, was impaired following knockdown of CaMKK or treatment with STO-609. Activation of CaMKK by BDNF requires transient receptor potential canonical (TRPC) channels as SKF-96365, but not the NMDA receptor antagonist d-APV, prevented BDNF-induced GluA1 surface expression as well as phosphorylation of CaMKI, AKT(T308), and mTOR. Using siRNA we confirmed the involvement of TRPC5 and TRPC6 subunits in BDNF-induced AKT(T308) phosphorylation. The BDNF-induced increase in mEPSC was blocked by IEM-1460, a selected antagonist of CP-AMPARs, as well as by the specific repression of acute GluA1 translation via siRNA to GluA1 but not GluA2. Together these data support the conclusion that newly synthesized GluA1 subunits, induced by BDNF, are readily incorporated into synapses where they enhance the expression of CP-AMPARs and synaptic strength.
Figures
References
-
- Aizenman CD, Muñoz-Elías G, Cline HT. Visually driven modulation of glutamatergic synaptic transmission is mediated by the regulation of intracellular polyamines. Neuron. 2002;34:623–634. - PubMed
-
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. - PubMed
-
- Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci. 2001;4:1093–1101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous