

Mitochondrial- and endoplasmic reticulum-associated oxidative stress in Alzheimer's disease: from pathogenesis to biomarkers
- PMID: 22701485
- PMCID: PMC3373122
- DOI: 10.1155/2012/735206
Mitochondrial- and endoplasmic reticulum-associated oxidative stress in Alzheimer's disease: from pathogenesis to biomarkers
Abstract
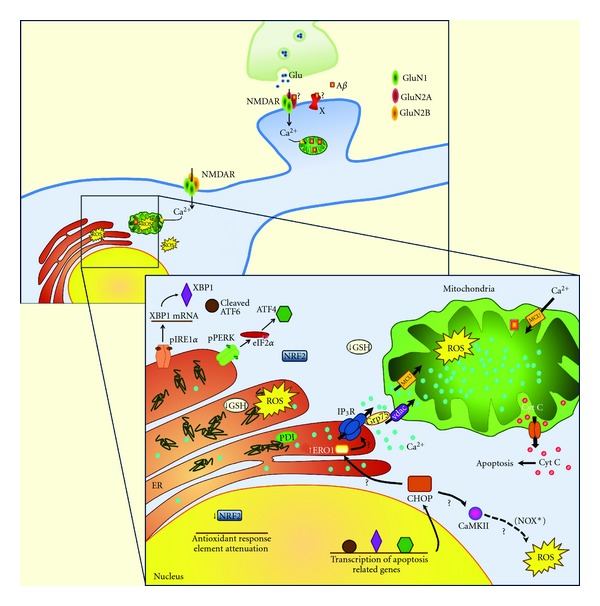
Alzheimer's disease (AD) is the most common cause of dementia in the elderly, affecting several million of people worldwide. Pathological changes in the AD brain include the presence of amyloid plaques, neurofibrillary tangles, loss of neurons and synapses, and oxidative damage. These changes strongly associate with mitochondrial dysfunction and stress of the endoplasmic reticulum (ER). Mitochondrial dysfunction is intimately linked to the production of reactive oxygen species (ROS) and mitochondrial-driven apoptosis, which appear to be aggravated in the brain of AD patients. Concomitantly, mitochondria are closely associated with ER, and the deleterious crosstalk between both organelles has been shown to be involved in neuronal degeneration in AD. Stimuli that enhance expression of normal and/or folding-defective proteins activate an adaptive unfolded protein response (UPR) that, if unresolved, can cause apoptotic cell death. ER stress also induces the generation of ROS that, together with mitochondrial ROS and decreased activity of several antioxidant defenses, promotes chronic oxidative stress. In this paper we discuss the critical role of mitochondrial and ER dysfunction in oxidative injury in AD cellular and animal models, as well as in biological fluids from AD patients. Progress in developing peripheral and cerebrospinal fluid biomarkers related to oxidative stress will also be summarized.
Figures
References
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. - PubMed
-
- Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39(3):409–421. - PubMed
-
- Miller DL, Papayannopoulos IA, Styles J, et al. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Archives of Biochemistry and Biophysics. 1993;301(1):41–52. - PubMed
-
- Teng CC, Yang YT, Chen YC, Kuo YM, Sze CI. Role of WWOX/WOX1 in Alzheimer's disease pathology and in cell death signaling. Frontiers in Bioscience. 2012;4:1951–1965. - PubMed
LinkOut - more resources
Full Text Sources
