

A highly scalable peptide-based assay system for proteomics
- PMID: 22701568
- PMCID: PMC3373263
- DOI: 10.1371/journal.pone.0037441
A highly scalable peptide-based assay system for proteomics
Abstract
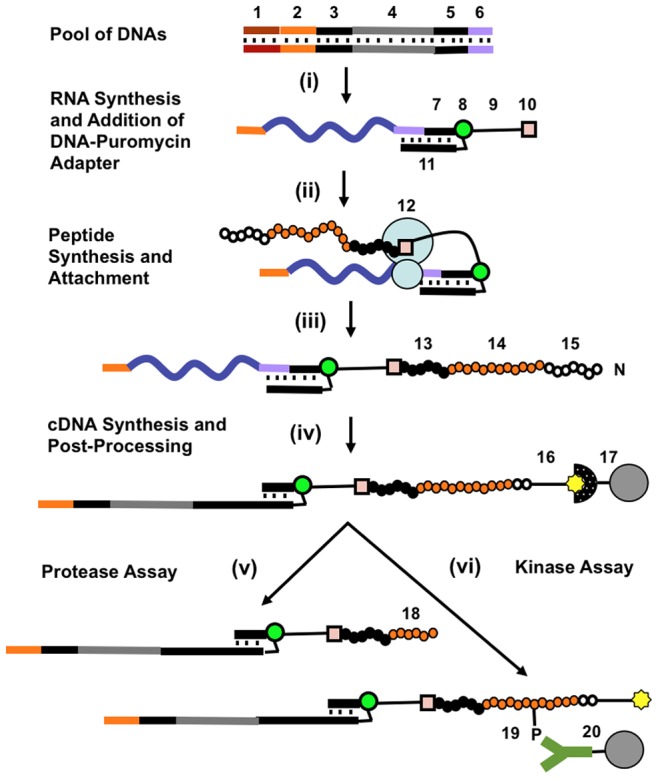
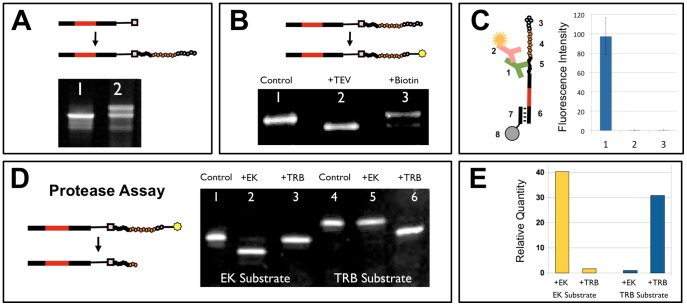
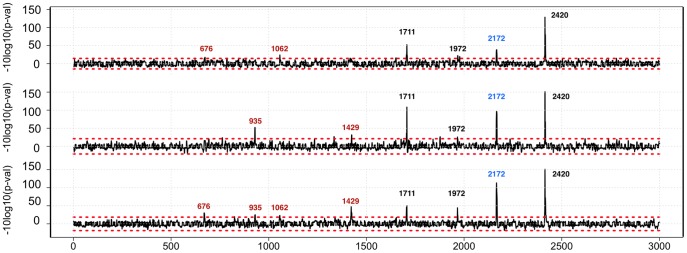
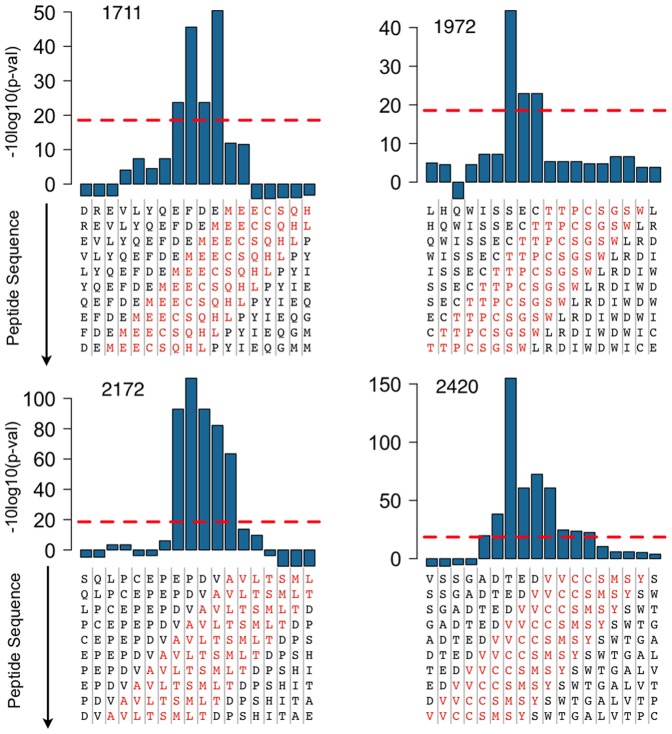
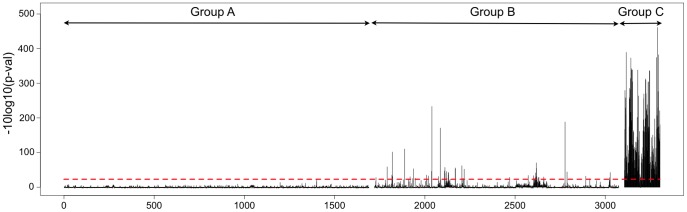
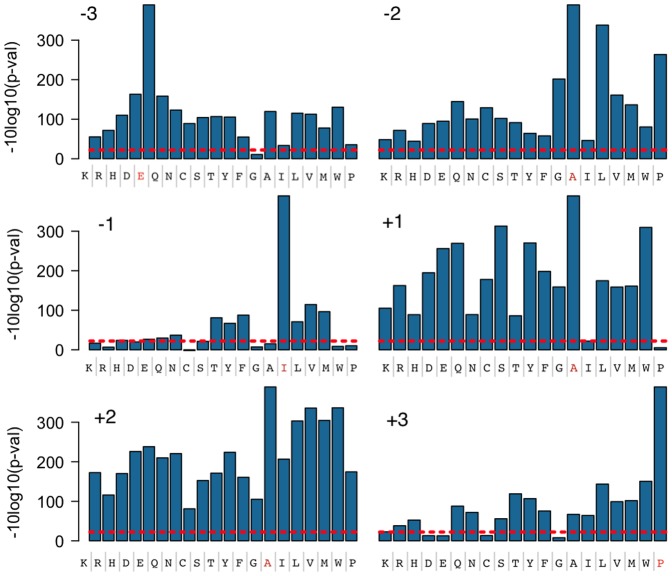
We report a scalable and cost-effective technology for generating and screening high-complexity customizable peptide sets. The peptides are made as peptide-cDNA fusions by in vitro transcription/translation from pools of DNA templates generated by microarray-based synthesis. This approach enables large custom sets of peptides to be designed in silico, manufactured cost-effectively in parallel, and assayed efficiently in a multiplexed fashion. The utility of our peptide-cDNA fusion pools was demonstrated in two activity-based assays designed to discover protease and kinase substrates. In the protease assay, cleaved peptide substrates were separated from uncleaved and identified by digital sequencing of their cognate cDNAs. We screened the 3,011 amino acid HCV proteome for susceptibility to cleavage by the HCV NS3/4A protease and identified all 3 known trans cleavage sites with high specificity. In the kinase assay, peptide substrates phosphorylated by tyrosine kinases were captured and identified by sequencing of their cDNAs. We screened a pool of 3,243 peptides against Abl kinase and showed that phosphorylation events detected were specific and consistent with the known substrate preferences of Abl kinase. Our approach is scalable and adaptable to other protein-based assays.
Conflict of interest statement
Figures
Similar articles
-
Quantitative proteomics identifies the membrane-associated peroxidase GPx8 as a cellular substrate of the hepatitis C virus NS3-4A protease.Hepatology. 2014 Feb;59(2):423-33. doi: 10.1002/hep.26671. Epub 2013 Dec 18. Hepatology. 2014. PMID: 23929719
-
Establishment of an in vitro assay system for screening hepatitis C virus protease inhibitors using high performance liquid chromatography.Antiviral Res. 1996 Aug;32(1):9-18. doi: 10.1016/0166-3542(95)00969-8. Antiviral Res. 1996. PMID: 8863991
-
How to find simple and accurate rules for viral protease cleavage specificities.BMC Bioinformatics. 2009 May 16;10:149. doi: 10.1186/1471-2105-10-149. BMC Bioinformatics. 2009. PMID: 19445713 Free PMC article.
-
Quantitative proteomics in plant protease substrate identification.New Phytol. 2018 May;218(3):936-943. doi: 10.1111/nph.14587. Epub 2017 May 11. New Phytol. 2018. PMID: 28493421 Review.
-
Methods for mapping protease specificity.Curr Opin Chem Biol. 2007 Feb;11(1):46-51. doi: 10.1016/j.cbpa.2006.11.021. Epub 2006 Dec 6. Curr Opin Chem Biol. 2007. PMID: 17157549 Review.
Cited by
-
Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with endemic human coronaviruses.Cell Rep Med. 2021 Jan 19;2(1):100189. doi: 10.1016/j.xcrm.2020.100189. Cell Rep Med. 2021. PMID: 33495758 Free PMC article.
-
COVID-19 vaccination elicits an evolving, cross-reactive antibody response to epitopes conserved with endemic coronavirus spike proteins.Cell Rep. 2022 Jul 5;40(1):111022. doi: 10.1016/j.celrep.2022.111022. Epub 2022 Jun 13. Cell Rep. 2022. PMID: 35753310 Free PMC article.
-
PepSeq as a highly multiplexed platform for melioidosis antigen discovery and vaccine development.Front Immunol. 2025 Jul 3;16:1605758. doi: 10.3389/fimmu.2025.1605758. eCollection 2025. Front Immunol. 2025. PMID: 40677719 Free PMC article.
-
Structural and functional diversity of metalloproteinases encoded by the Bacteroides fragilis pathogenicity island.FEBS J. 2014 Jun;281(11):2487-502. doi: 10.1111/febs.12804. Epub 2014 Apr 22. FEBS J. 2014. PMID: 24698179 Free PMC article.
-
High-resolution analysis and functional mapping of cleavage sites and substrate proteins of furin in the human proteome.PLoS One. 2013;8(1):e54290. doi: 10.1371/journal.pone.0054290. Epub 2013 Jan 15. PLoS One. 2013. PMID: 23335997 Free PMC article.
References
-
- Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Large-Scale Genome Sequencing Program. National Human Genome Research Institute website. 2011;24 Available: www.genome.gov/sequencingcosts Accessed 2012 April.
-
- Scholle MD, Kriplani U, Pabon A, Sishtla K, Glucksman MJ, et al. Mapping protease substrates by using a biotinylated phage substrate library. Chembiochem. 2006;7:834–838. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
