

Identification and validation of copy number variants using SNP genotyping arrays from a large clinical cohort
- PMID: 22702538
- PMCID: PMC3464625
- DOI: 10.1186/1471-2164-13-241
Identification and validation of copy number variants using SNP genotyping arrays from a large clinical cohort
Abstract
Background: Genotypes obtained with commercial SNP arrays have been extensively used in many large case-control or population-based cohorts for SNP-based genome-wide association studies for a multitude of traits. Yet, these genotypes capture only a small fraction of the variance of the studied traits. Genomic structural variants (GSV) such as Copy Number Variation (CNV) may account for part of the missing heritability, but their comprehensive detection requires either next-generation arrays or sequencing. Sophisticated algorithms that infer CNVs by combining the intensities from SNP-probes for the two alleles can already be used to extract a partial view of such GSV from existing data sets.
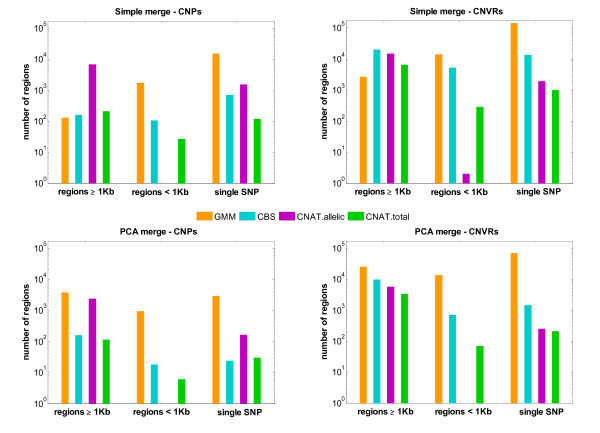
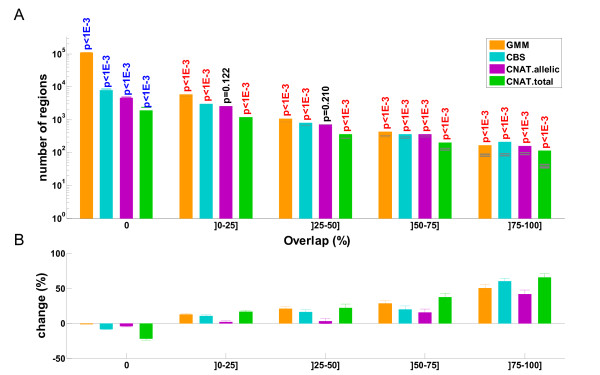
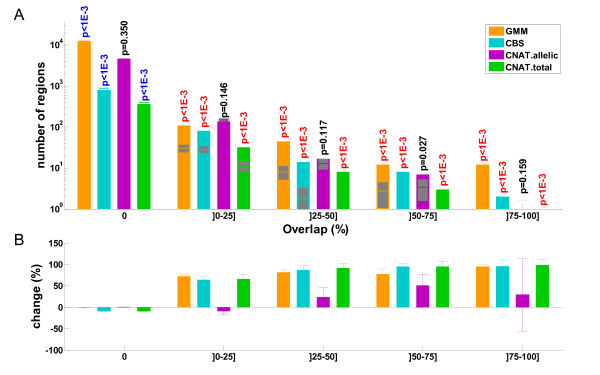
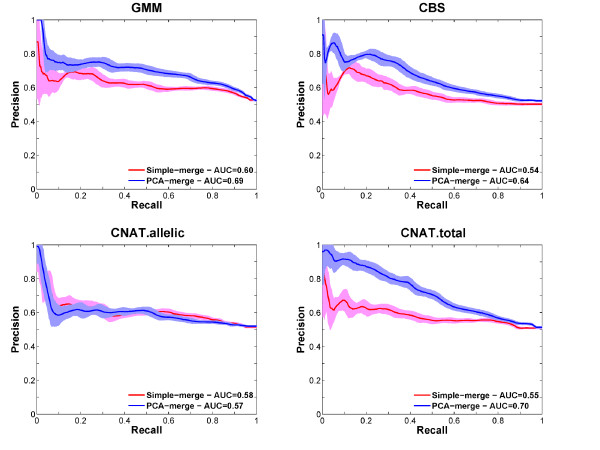
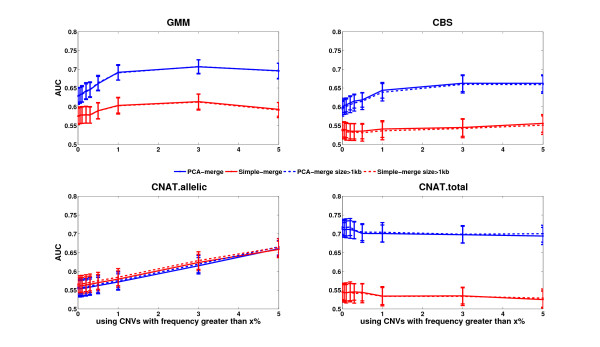
Results: Here we present several advances to facilitate the latter approach. First, we introduce a novel CNV detection method based on a Gaussian Mixture Model. Second, we propose a new algorithm, PCA merge, for combining copy-number profiles from many individuals into consensus regions. We applied both our new methods as well as existing ones to data from 5612 individuals from the CoLaus study who were genotyped on Affymetrix 500K arrays. We developed a number of procedures in order to evaluate the performance of the different methods. This includes comparison with previously published CNVs as well as using a replication sample of 239 individuals, genotyped with Illumina 550K arrays. We also established a new evaluation procedure that employs the fact that related individuals are expected to share their CNVs more frequently than randomly selected individuals. The ability to detect both rare and common CNVs provides a valuable resource that will facilitate association studies exploring potential phenotypic associations with CNVs.
Conclusion: Our new methodologies for CNV detection and their evaluation will help in extracting additional information from the large amount of SNP-genotyping data on various cohorts and use this to explore structural variants and their impact on complex traits.
Figures
References
-
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. - PubMed
-
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. - PubMed
-
- Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D. et al.Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
