

Detection of horizontal transfer of individual genes by anomalous oligomer frequencies
- PMID: 22702893
- PMCID: PMC3497702
- DOI: 10.1186/1471-2164-13-245
Detection of horizontal transfer of individual genes by anomalous oligomer frequencies
Abstract
Background: Understanding the history of life requires that we understand the transfer of genetic material across phylogenetic boundaries. Detecting genes that were acquired by means other than vertical descent is a basic step in that process. Detection by discordant phylogenies is computationally expensive and not always definitive. Many have used easily computed compositional features as an alternative procedure. However, different compositional methods produce different predictions, and the effectiveness of any method is not well established.
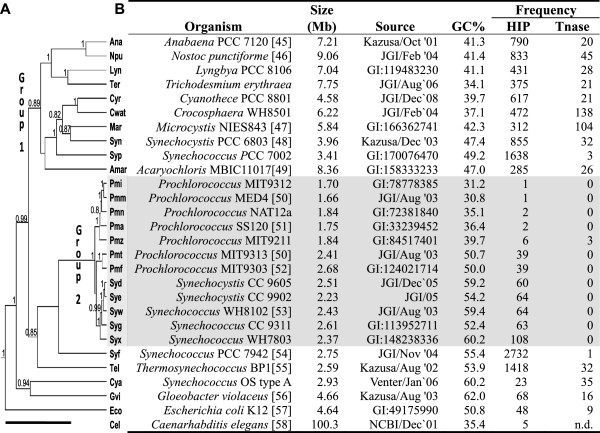
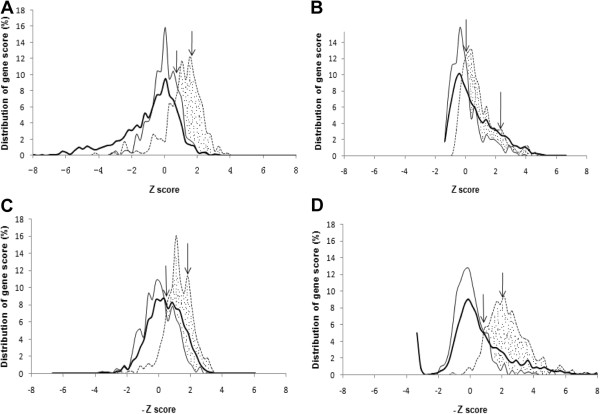
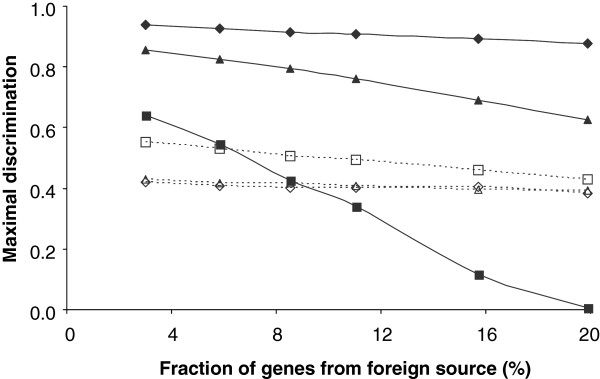
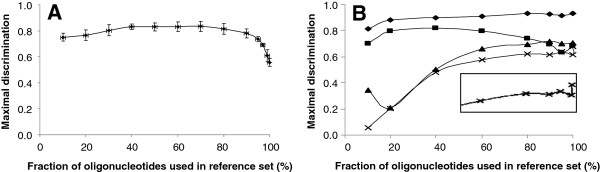
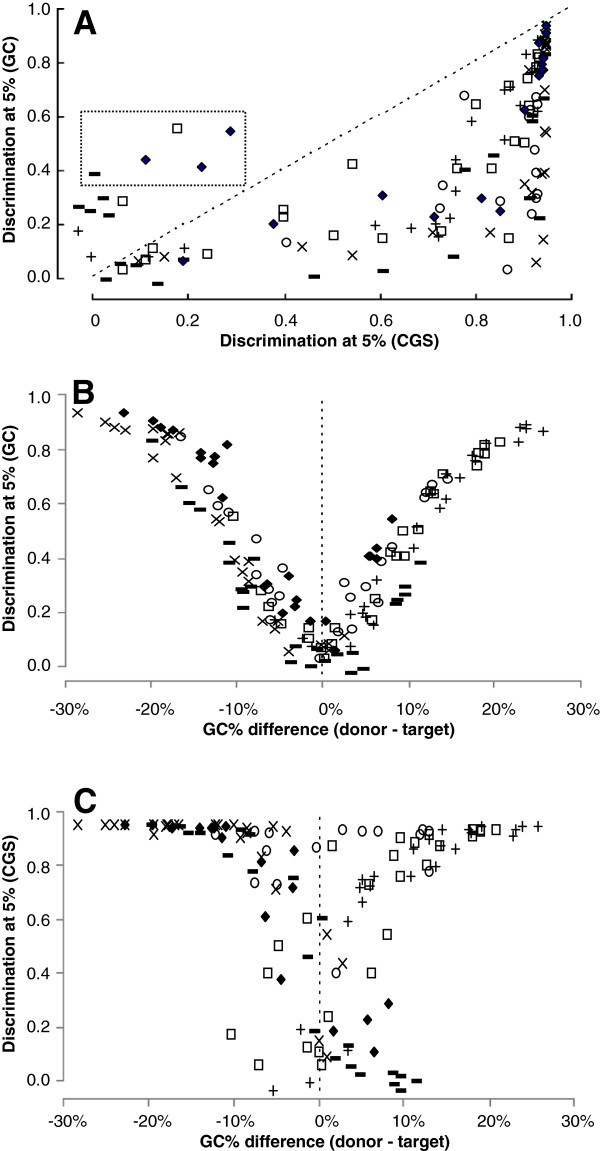
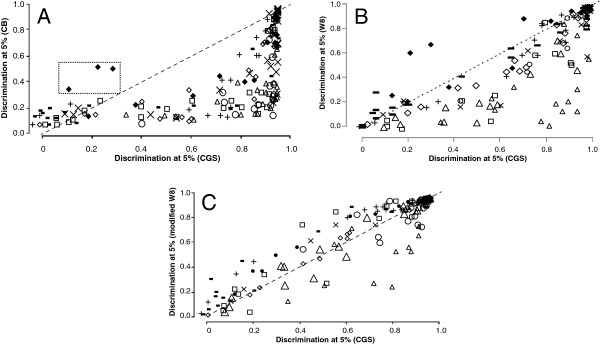
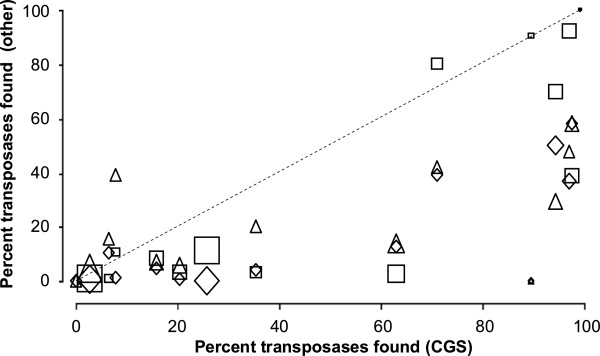
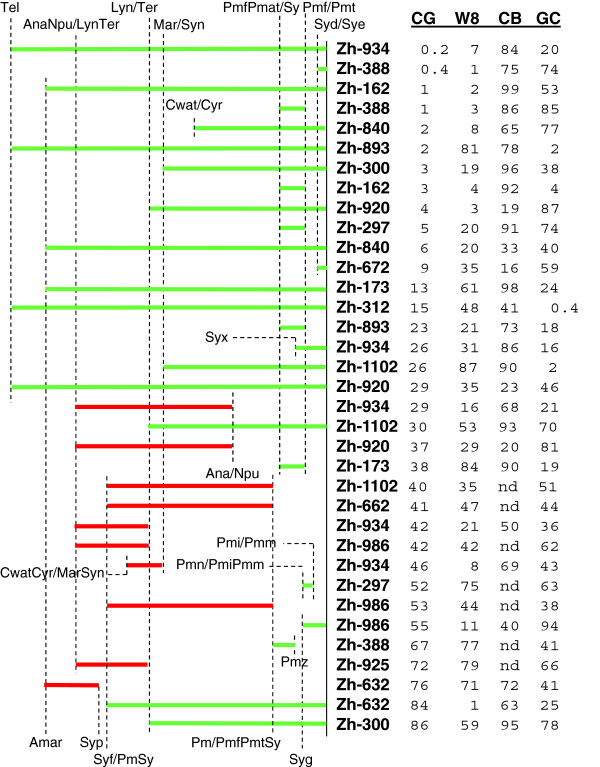
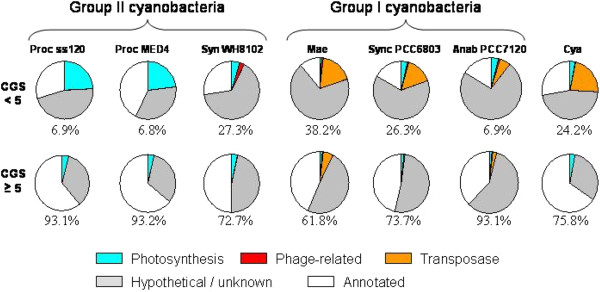
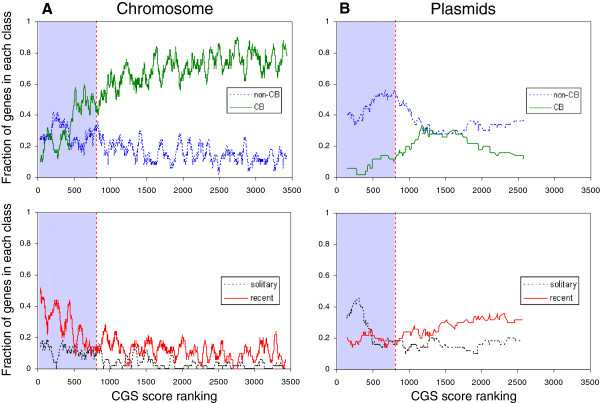
Results: The ability of octamer frequency comparisons to detect genes artificially seeded in cyanobacterial genomes was markedly increased by using as a training set those genes that are highly conserved over all bacteria. Using a subset of octamer frequencies in such tests also increased effectiveness, but this depended on the specific target genome and the source of the contaminating genes. The presence of high frequency octamers and the GC content of the contaminating genes were important considerations. A method comprising best practices from these tests was devised, the Core Gene Similarity (CGS) method, and it performed better than simple octamer frequency analysis, codon bias, or GC contrasts in detecting seeded genes or naturally occurring transposons. From a comparison of predictions with phylogenetic trees, it appears that the effectiveness of the method is confined to horizontal transfer events that have occurred recently in evolutionary time.
Conclusions: The CGS method may be an improvement over existing surrogate methods to detect genes of foreign origin.
Figures
Similar articles
-
Phylogenetic analyses of cyanobacterial genomes: quantification of horizontal gene transfer events.Genome Res. 2006 Sep;16(9):1099-108. doi: 10.1101/gr.5322306. Epub 2006 Aug 9. Genome Res. 2006. PMID: 16899658 Free PMC article.
-
Horizontal gene transfer in cyanobacterial signature genes.Methods Mol Biol. 2009;532:339-66. doi: 10.1007/978-1-60327-853-9_20. Methods Mol Biol. 2009. PMID: 19271195 Review.
-
[Phylogenetic application and analysis of horizontal transfer based on the prokaryote eno gene].Yi Chuan. 2012 Jul;34(7):907-18. doi: 10.3724/sp.j.1005.2012.00907. Yi Chuan. 2012. PMID: 22805218 Chinese.
-
The plastid ancestor originated among one of the major cyanobacterial lineages.Nat Commun. 2014 Sep 15;5:4937. doi: 10.1038/ncomms5937. Nat Commun. 2014. PMID: 25222494
-
Phylogenomic networks.Trends Microbiol. 2011 Oct;19(10):483-91. doi: 10.1016/j.tim.2011.07.001. Epub 2011 Aug 3. Trends Microbiol. 2011. PMID: 21820313 Review.
Cited by
-
SWPhylo - A Novel Tool for Phylogenomic Inferences by Comparison of Oligonucleotide Patterns and Integration of Genome-Based and Gene-Based Phylogenetic Trees.Evol Bioinform Online. 2018 Feb 20;14:1176934318759299. doi: 10.1177/1176934318759299. eCollection 2018. Evol Bioinform Online. 2018. PMID: 29511354 Free PMC article.
-
Alignment-free inference of hierarchical and reticulate phylogenomic relationships.Brief Bioinform. 2019 Mar 22;20(2):426-435. doi: 10.1093/bib/bbx067. Brief Bioinform. 2019. PMID: 28673025 Free PMC article. Review.
-
Microbial genomic island discovery, visualization and analysis.Brief Bioinform. 2019 Sep 27;20(5):1685-1698. doi: 10.1093/bib/bby042. Brief Bioinform. 2019. PMID: 29868902 Free PMC article. Review.
-
Computational methods for predicting genomic islands in microbial genomes.Comput Struct Biotechnol J. 2016 May 7;14:200-6. doi: 10.1016/j.csbj.2016.05.001. eCollection 2016. Comput Struct Biotechnol J. 2016. PMID: 27293536 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
